CpH MD Tutorial
Based upon the 2009 CTBP Workshop tutorial. The tutorial files have some mistakes in them, and we don't use PBS queuing, which changes some nuts and bolts. It is still a good idea to download and run through that tutorial, as it uses CHARMM at least in the early stages, and sometimes CHARMM does things a bit differently than when being called through MMTSB.
There is also useful information at this (continuous-constant pH) tutorial, especially on time-based convergence. We will use 2EVO and our CE8K12 peptide (the titratable residues are GLU and LYS). Later, we can use CE8K12 peptide for our CYS analysis.
FILES Here.
In order to make the preparation as simple as possible, we will pass as many of preparation steps directly to mdCHARMM.pl/aarex.pl. This includes replacing/fixing histidine residues, patching any other titratable residues, and referencing pHMD topology and structure files.
The general structure of data directories should be to separate structure/input files from data. Although this is a bit confusing and requires planning at first, it makes it much easier to organize and find data later. Remember, you will often come back to data months or years later with the need to make sense of it.
Single pH MD
Preparing the structure
Converting HIS residues can be done simply with convpdb.pl. We also make sure the PDB will be in CHARMM22 format. The "see" bit utilizes regular expression substitution. We are replacing occurrences of "HSD" with "HSP". HSP is the protonated HST residue.
$ convpdb.pl -out charmm22 -segnames PROTEIN.pdb | sed "s/HSD/HSP/g" > peptide.pdb
If your PDB file is already "charmm clean", you don't have to throw the "-out charmm22" switch. At this point, you can use MMTSB to add any hydrogens, generate a PSF and/or CRD file, and so on.
Test: Single-temperature CpH MD
As a beginning test of CpHMD, we may run a single-temperature MD simulation, which will record the λ values for each titratable group per output time step. Notice that all the patches are applied right in the mdCHARMM command. Solvated systems will need the salt concentration specified; this is set to be 0.15M and the information is read as a custom file (setup.str)
Files: md-cphmd-ce8k12-ph3.bash (input); xxx.dcd, lambda data, logfile (output)
Note that you will probably have to generate a PSF file. While it is convenient to not have to input a PSF file for the simulation (MMTSB generates one on the fly), it is important to realize that for this simulation (and the REX simulations, below) that the input PDB file is patched, which changes the atom types and total number in the molecule. When you generate a PSF from the initial PDB, you must follow the same patching or else you will get a mismatch. In addition, the correct topology files must be specified. A sample command is contained in each script for convenience.
T-based Replica Exchange CpH MD
Replica exchange allows us to explore more of the simulation space, by exchanging replicas amongst different temperatures. This enhanced sampling will allow more confident determination of pKa values, and also allow the calculation of potentials of mean force (PMF). Although many times you will see REX simulations implemented through a queue (PBS/qsub) or MPI (mpirun), for our purposes, running in "dumb" mode works well (on sierra or on an OS X machine).
files: PSFrex-ce8k12-ph3.bash, mpirun-run1.ph3.bash, rex-cphmd-2evq-ph1.5.bash, rex-cphmd-ce8k12-ph3.bash (input)
These files can be edited to change the pH and saved so you may even make a "script of scripts" or run them sequentially.
Start with rex-cphmd-2evq-ph1.5.bash or rex-cphmd-ce8k12-ph3.bash.
Analysis
Analysis follows the CPBT tutorial in a straightforward manner.
pKa/protonated fraction analysis
The general flow of data processing is:
The first step could be scripted, so you could generate all the .lambda and .sx files at once. See gen-pKa-ens_all.csh which can be used as a template to utilize many pH/replica points for the S curves for the generation of pKa/S curves. Note that by default, cptpKa.pl will send the PostScript plots to your printer; this is a bit inconvenient especially if you don't have your default printer connected. (recall that you may use the UNIX command 'lpq' to see if there are any jobs stuck in the default printer queue, and 'lprm' to delete any such jobs).
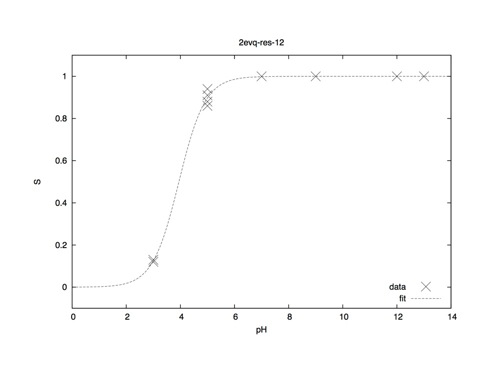
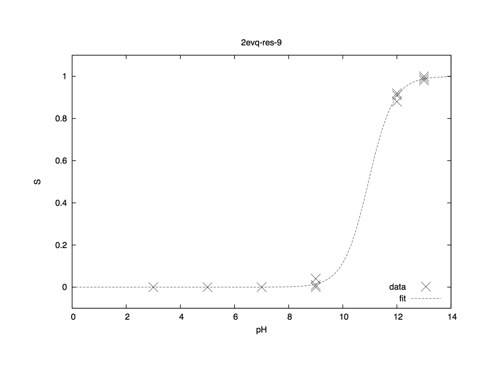
Utilizing 4 replicas at each pH point, data should look something like this (per residue; one GLU and one LYS contained in 2EVQ).
Trajectory/structure analysis
In order to obtain trajectories of the replica exchange data, you will have to use rexanalysis.pl.
New titratable residues
We want to investigate the thiol proton acidity in CYS. A bare thiol proton in CYS has pKa = 8.3 or so. Since the current version of CHARMM (c38) does not have parameters for CYS, we have to develop the parameters that pHMD will use for CYS. The pertinent documentation is here (Examples) and in the CTBP Workshop Tutorial (Step 5).
Start with a simple, standalone CYS (blocked at the ends), prepare the simulations as described, and extract the PMF and parameters. Next, run REX-CPHMD to confirm an accurate pKa. The next step will be larger CYS-containing peptides and proteins.
Based upon the 2009 CTBP Workshop tutorial. The tutorial files have some mistakes in them, and we don't use PBS queuing, which changes some nuts and bolts. It is still a good idea to download and run through that tutorial, as it uses CHARMM at least in the early stages, and sometimes CHARMM does things a bit differently than when being called through MMTSB.
There is also useful information at this (continuous-constant pH) tutorial, especially on time-based convergence. We will use 2EVO and our CE8K12 peptide (the titratable residues are GLU and LYS). Later, we can use CE8K12 peptide for our CYS analysis.
FILES Here.
In order to make the preparation as simple as possible, we will pass as many of preparation steps directly to mdCHARMM.pl/aarex.pl. This includes replacing/fixing histidine residues, patching any other titratable residues, and referencing pHMD topology and structure files.
The general structure of data directories should be to separate structure/input files from data. Although this is a bit confusing and requires planning at first, it makes it much easier to organize and find data later. Remember, you will often come back to data months or years later with the need to make sense of it.
Single pH MD
Preparing the structure
Converting HIS residues can be done simply with convpdb.pl. We also make sure the PDB will be in CHARMM22 format. The "see" bit utilizes regular expression substitution. We are replacing occurrences of "HSD" with "HSP". HSP is the protonated HST residue.
$ convpdb.pl -out charmm22 -segnames PROTEIN.pdb | sed "s/HSD/HSP/g" > peptide.pdb
If your PDB file is already "charmm clean", you don't have to throw the "-out charmm22" switch. At this point, you can use MMTSB to add any hydrogens, generate a PSF and/or CRD file, and so on.
Test: Single-temperature CpH MD
As a beginning test of CpHMD, we may run a single-temperature MD simulation, which will record the λ values for each titratable group per output time step. Notice that all the patches are applied right in the mdCHARMM command. Solvated systems will need the salt concentration specified; this is set to be 0.15M and the information is read as a custom file (setup.str)
Files: md-cphmd-ce8k12-ph3.bash (input); xxx.dcd, lambda data, logfile (output)
Note that you will probably have to generate a PSF file. While it is convenient to not have to input a PSF file for the simulation (MMTSB generates one on the fly), it is important to realize that for this simulation (and the REX simulations, below) that the input PDB file is patched, which changes the atom types and total number in the molecule. When you generate a PSF from the initial PDB, you must follow the same patching or else you will get a mismatch. In addition, the correct topology files must be specified. A sample command is contained in each script for convenience.
T-based Replica Exchange CpH MD
Replica exchange allows us to explore more of the simulation space, by exchanging replicas amongst different temperatures. This enhanced sampling will allow more confident determination of pKa values, and also allow the calculation of potentials of mean force (PMF). Although many times you will see REX simulations implemented through a queue (PBS/qsub) or MPI (mpirun), for our purposes, running in "dumb" mode works well (on sierra or on an OS X machine).
files: PSFrex-ce8k12-ph3.bash, mpirun-run1.ph3.bash, rex-cphmd-2evq-ph1.5.bash, rex-cphmd-ce8k12-ph3.bash (input)
These files can be edited to change the pH and saved so you may even make a "script of scripts" or run them sequentially.
Start with rex-cphmd-2evq-ph1.5.bash or rex-cphmd-ce8k12-ph3.bash.
Analysis
Analysis follows the CPBT tutorial in a straightforward manner.
pKa/protonated fraction analysis
The general flow of data processing is:
- Create a lambda and Sx data file for each replica (using getRexLam.csh, note we bypass using extractLamb.pl and cptSX.pl)
- Calculate pKa values and fit to Henderson-Hasselbach (using cptpKa.pl)
The first step could be scripted, so you could generate all the .lambda and .sx files at once. See gen-pKa-ens_all.csh which can be used as a template to utilize many pH/replica points for the S curves for the generation of pKa/S curves. Note that by default, cptpKa.pl will send the PostScript plots to your printer; this is a bit inconvenient especially if you don't have your default printer connected. (recall that you may use the UNIX command 'lpq' to see if there are any jobs stuck in the default printer queue, and 'lprm' to delete any such jobs).
Utilizing 4 replicas at each pH point, data should look something like this (per residue; one GLU and one LYS contained in 2EVQ).
Trajectory/structure analysis
In order to obtain trajectories of the replica exchange data, you will have to use rexanalysis.pl.
New titratable residues
We want to investigate the thiol proton acidity in CYS. A bare thiol proton in CYS has pKa = 8.3 or so. Since the current version of CHARMM (c38) does not have parameters for CYS, we have to develop the parameters that pHMD will use for CYS. The pertinent documentation is here (Examples) and in the CTBP Workshop Tutorial (Step 5).
Start with a simple, standalone CYS (blocked at the ends), prepare the simulations as described, and extract the PMF and parameters. Next, run REX-CPHMD to confirm an accurate pKa. The next step will be larger CYS-containing peptides and proteins.