First we want to prepare some structures. Here, we illustrate how to prepare ten independent proline 8-mer structures, each in its own
directory (note that after you type in the first "while", the prompt changes to "while?"; you exit this while loop with the "end" command): REMEMBER that you must have your little file with the one-letter sequence (pro8.one, though it doesn't really matter exactly what sequence you create or use for this illustration!).
$ genseq.pl -one pro8.one > pro8.seq
$ set suff=1
$ set filename=pro8
$ set nres=8
$ set mdpar="-par cmap,blocked"
$ while ( $suff < = 10 )
while? echo "loop" $suff
while? mkdir $filename.$suff
while? cd $filename.$suff
while? genchain.pl -g 100 -rnd $nres > $filename.chain
while? rebuild.pl ../$filename.seq $filename.chain > $filename.ca.pdb
while? minCHARMM.pl $mdpar $filename.ca.pdb > $filename.min.pdb
while? @ suff = $suff + 1
while? cd ../
while? end
$ genPSF.pl $mdpar $filename.1/$filename.min.pdb > $filename.psf
After these steps, the $suff counter is 11, and if you have to re-run because of some error, the while loop will fail. In that case, you must reset the $suff counter to 1.
You will make mistakes (typos) typically when doing this the first time. If your little script fails, then start again from the first "set suff = 1" command. Typically, once you have something like this working, you copy all the lines and put them into a file, which you then modify and run as necessary (so you don't keep making typos!. The entire script has to start with a special command. Here is the whole thing:
#!/bin/csh -f
set suff=1
set filename=pro8
set nres=8
set mdpar="-par cmap,blocked"
while ( $suff < 10 )
echo "loop" $suff
mkdir $filename.$suff
cd $filename.$suff
genchain.pl -g 100 -rnd $nres > $filename.chain
rebuild.pl ../$filename.seq $filename.chain > $filename.ca.pdb
minCHARMM.pl $mdpar $filename.ca.pdb > $filename.min.pdb
@ suff = $suff + 1
cd ..
end
If you copy all this into a file (say, script.csh), you have to tell the computer it is considered a program in order to use it. That is accomplished bye the following command:
chmod 755 script.csh
In order to run it, just type it's name:
./script.csh
All of this (except maybe the use of the while loop) should be clear to you at this point. We will utilize these different structures later on in the course. Right now, we want
to generate some data from a single structure. Let's simply use the first one, contained in the directory pro8.1/ for now. Change into that directory and proceed.
Notice the differences in energy between starting structure and the minimized structure. If you apply further minimization to the minimized structure, you can get a feel for how much minimization is needed. The MMTSB documentation states:
Without any options a 50-step steepest descent minimization is followed by the more aggressive adopted-basis Newton-Raphson (ABNR) minimization algorithm over 500 steps or until the energy decrease between steps becomes less than 1.0E-5 kcal/mol. With the default protocol the minimization is carried out with the CHARMM22 protein force field in vacuum (constant dielectric of 1.0) with a 14.0 A cutoff for non-bonded interactions.
If you examine the files before and after minimization, you will see the coordinates have changed:
$ more pro8.ca.pdb
$ more pro8.min.pdb
Now check the energies:
$ enerCHARMM.pl -par cmap,blocked pro8.ca.pdb
19523540364.0000
$ enerCHARMM.pl -par cmap,blocked pro8.min.pdb
308.0082
$ minCHARMM.pl -par cmap,blocked pro8.min.pdb > pro8.min2.pdb
$ enerCHARMM.pl -par cmap,blocked pro8.min2.pdb
301.4959
In order to begin to aquire data, we use the mdCHARMM.pl command. This command is able to take MANY arguments. To begin, we will heat the structure from 0 to 300 K in steps of 50 K. We create both a CHARMM energy log file and an MMTSB-type energy log file. Finally, we save the last configuration as ...final.pdb.
$ mdCHARMM.pl pro8.min.pdb -enerout pro8.heat.out -elog pro8.elog.out \
-trajout pro8.heat.dcd -log charmm-heat.log \
-par cmap,blocked,dynsteps=7000,dyndeltat=50.0,dynhtfrq=1000,dynitemp=0 \
-final pro8.T300.final.pdb
This generates the data we need in order to analyze whether our simulation is doing what it should. The first hurdle we have to get over is how to look at this data. If you examine pro8.heat.out, the CHARMM file which contains all the temperature and energy data, it consists of a header block, followed by a block of data for each timestep which is written out (default every 50 timesteps):
5 4 -33
STEP TIME TOTE TOTK ENER
EP-K TEMP BOND ANGL DIHE
IMPR VDW ELEC HBON HARM
DMC RGY
50 0.1000 308.1199 0.2495 307.8705
307.6210 0.8287 8.2777 56.3792 79.1510
0.2102 4.7890 157.9107 0.0000 0.0000
0.0000 0.0000
The CHARMM file header block is nice because it tells you what each number is, but the format of the file itself is not very compact. The file pro8.elog.out is a little better; it gives mostly the same information, but you may need to look at the CHARMM header to remember what each column is:
MD: 0 0.00 0.00 308.12 0.00 308.12 5.15 157.96 0.00 0.00 0.00 0.000000 0.000000
MD: 50 0.10 0.83 308.12 0.25 307.87 4.79 157.91 0.00 0.00 0.00 0.000000 0.000000
This data can be plotted by most plotting programs without difficulty.
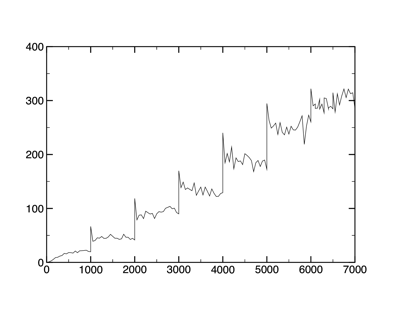
MMTSB-type data (Energy versus Temperature)
The third way to extract data is to extract pertinent lines the CHARMM output file. This is kind of messy, but gives the most complete set of information. In order to do this, I have written a script which makes the necessary extractions, makedata.csh. You can edit this script to match the output file name (or make your CHARMM log file match the name in the script in this example it's "ht.out"), make the script executable, and run it:
$ chmod 755 makedata.csh
$ ./makedata.csh
$ ls -ls
8 -rw-r--r-- 1 mccallum staff 4056 Oct 12 10:37 data-presscp.dat
8 -rw-r--r-- 1 mccallum staff 4056 Oct 12 10:37 data-pecp.dat
8 -rw-r--r-- 1 mccallum staff 3588 Oct 12 10:37 data-T.dat
These files contain pressure, potential energy, and temperature data, respectively. Note, however that this has not exactly done the timesteps right; in other words, it is reporting time, not timesteps.
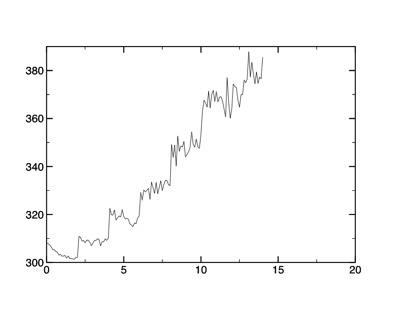
Energy data obtained from the makedata.csh script.
What do these data tell us? Looking at the temperature data, you can clearly see each temperature jump (every 1,000 steps). The temperature of the simulation appears to be increasing and stabilizing 50 K at a time. Or is it? Notice also that as the temperature rises, the fluctuations increase.
The energy is another matter. Notice that the potential energy does not look at all as "clean" as the temperature data. You must pay careful attention to the energy, as it will show when you can expect problems.
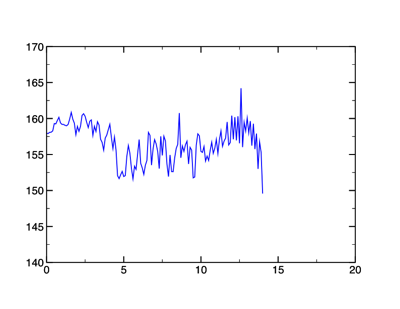
Potential energy (kcal/mol) versus time (ps)
To investigate this further, in the next lecture, we will heat, equilibrate and run "production" MD. What is the difference?
- Heating: the system is allowed to exchange (usually absorb) energy with an outside source ("bath") in the form of kinetic energy. Since temperature can be measured through kinetic energy (and vice-versa), the system is progressively injected with more energy (temperature) in stages, from 0K to 300K or so.
- Equilibration: the system is allowed to exchange energy with the bath, but at a constant measured temperature. If the system cools (as measured by the kinetic energy), more energy is added. If it heats, some is allowed to drain. This continues until the temperature is constant within fluctuations.
- Production: the system is NOT allowed to exchange energy with the bath, so energy is conserved within the system. Any slope in the energy versus time at this point means the systems has not been properly equilibrated.