The common understanding of post-ionization fragmentation of molecules is that any excess
electronic energy left in the molecule is quickly converted to vibrational energy, which
then dissociates along different coordinates in a purely statistical manner,
controlled by transition-state theory.
Although the possibilities of other mechanisms have been acknowleged (roaming, impulsive
dissociation), these are computationally demanding to simulate.
In particular, the electronic states of these open-shell species are complex, especially
as bonds also break.
In our work, this is compounded when a dissociation furthermore takes place on
an excited-state surface.
We have done a mixture of intense computational work and reductive theoretical distillation
to understand impulsive, excited-state dissociations of molecular ions.
Project publications. Click sidebar to see all publications.
|
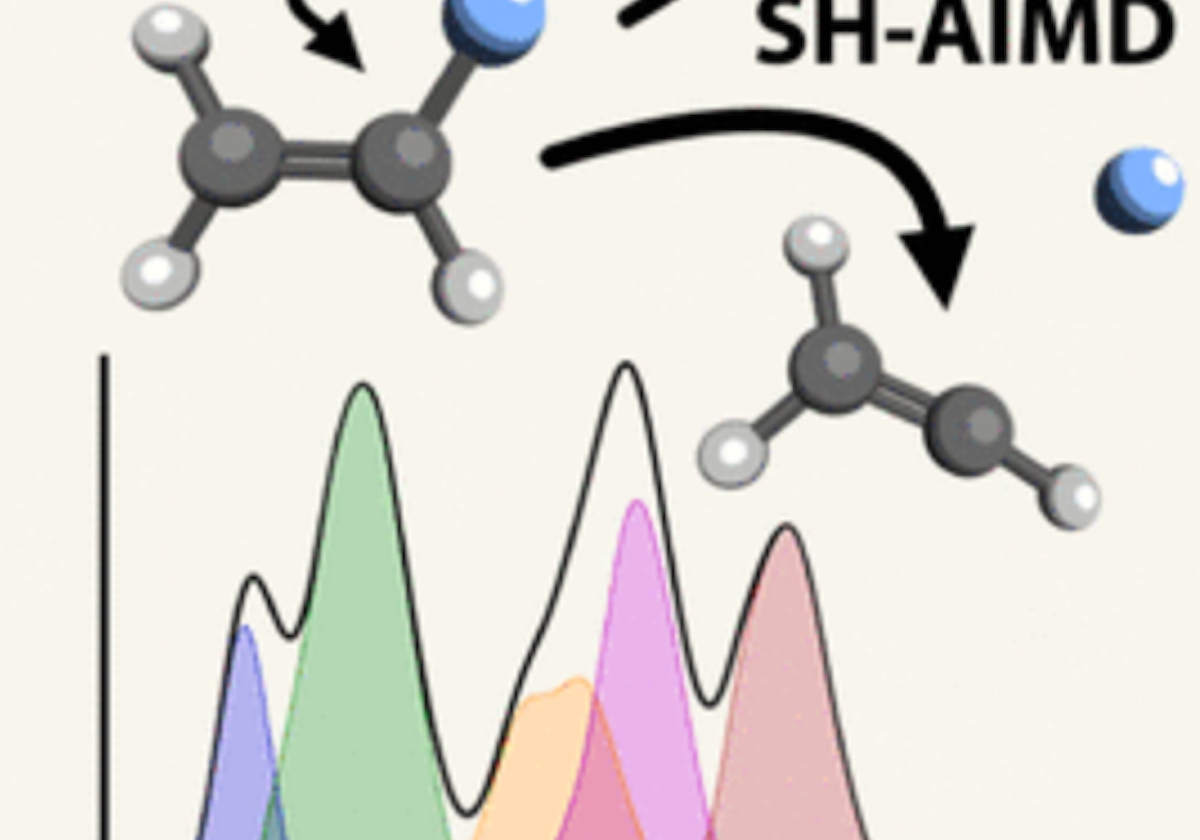
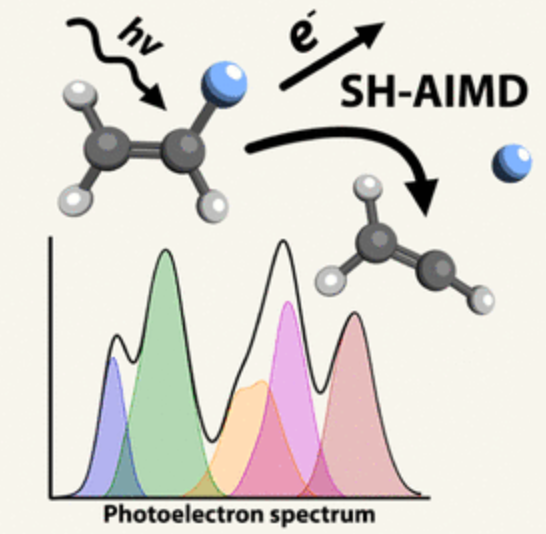
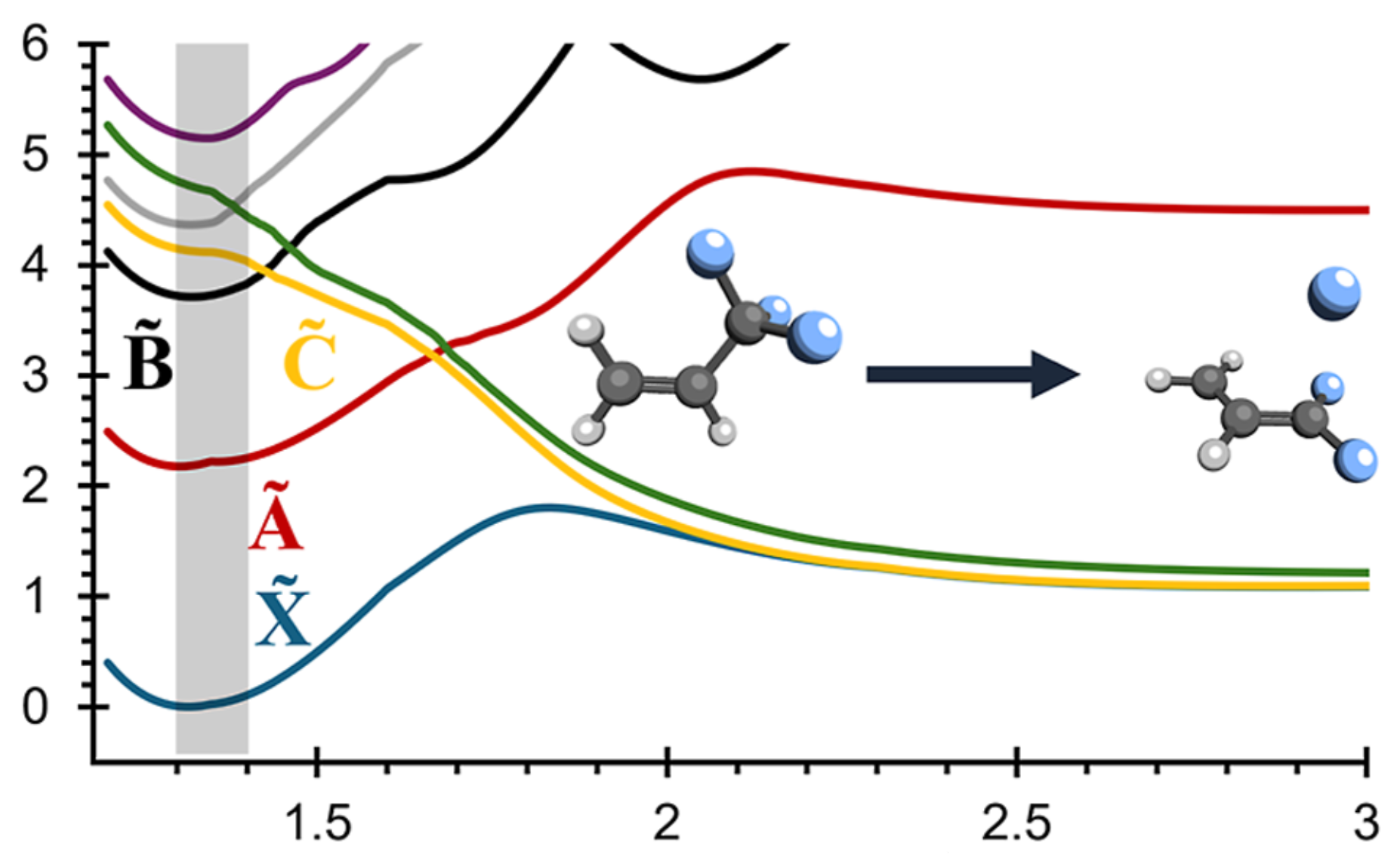
Long-standing debates regarding the dissociative photoionization of vinyl fluoride (fluoroethene) were resolved using large-scale surface-hopping ab initio molecular dynamics (SH-AIMD) simulations. By combining accurate initial condition sampling, electronic cross-section calculations, and SH-AIMD with density functional theory (DFT) and complete active space second-order perturbation theory (CASPT2), we obtained not only qualitative insight into excited-state dynamics but also quantitatively accurate predictions of the photoelectron spectrum, fluorine-loss branching ratios, and translational kinetic energy release distributions for F + C2H3+ products. Statistical dissociation arises from the X̃ 2A″–B̃ 2A′ states, while, in the C̃ 2A″–Ẽ 2A′ states, excited-state dissociation within 50–250 fs dominates. Only CASPT2 captures the formation of an excited triplet C2H3+ fragment, though DFT still reproduces correct branching ratios, as branching pathways are largely determined at short bond distances. Importantly, the previously hypothesized inclusion of autoionizing Rydberg states is not required to match experimental observables.
|
|
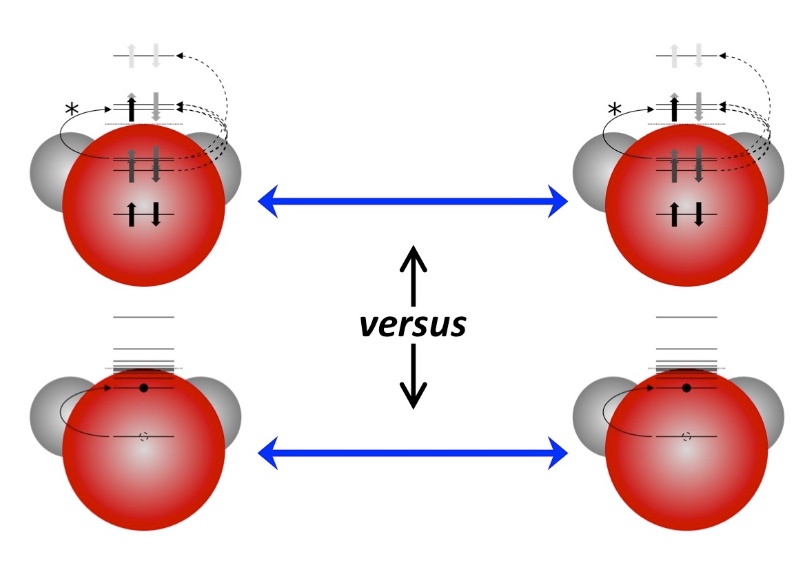
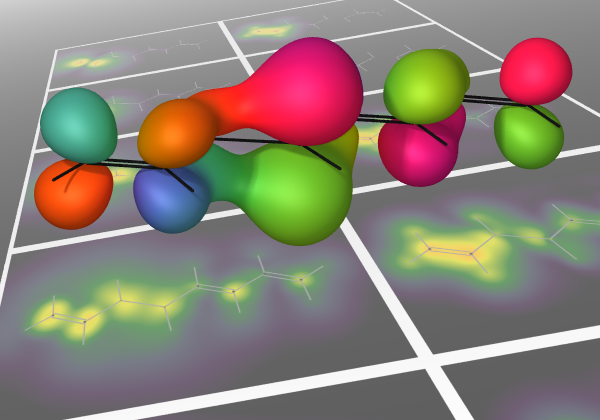
Valence photoionization generates photoions with excess internal energy, often resulting in statistical dissociation, well described by RRKM theory. Yet, numerous small photoions with electronegative substituents (e.g., F, Cl, Br, OH) have been shown to exhibit nonstatistical behavior, undergoing direct dissociation from repulsive electronic states to yield radicals such as F• or OH•, along with their corresponding fragment ions. Here, we present a general, predictive model that rationalizes this mechanism and extends it to molecules bearing electronegative substituents capable of forming 2P (F•, Cl•, Br•) or 2Π (OH•, SH•, N3•, NCO•) radicals. Nonstatistical dissociation arises from ionization of p- or π-localized orbitals on electronegative atoms, producing radical-like fragments unbound to the cationic core. The outcome is governed by three factors: excitation energy, the bonding character of the ionized orbital, and electronic degeneracy between states upon dissociation. Using these criteria, we define four classes of potential energy surfaces, enabling predictive classification of dissociative behavior. Extensive computations on over 50 molecules confirm the model's accuracy and generality.
|
|
 go to UOP homepage
go to UOP homepage
 © Copyright 2012–2026
© Copyright 2012–2026