Real-time quantum dynamics studies are already a mainstay for nuclear motions of complex systems.
Dynamic studies are necessary because the density of states at relevant energies
renders the eigenstate formulation computationally intractable.
We should not expect the situation to be any different as we consider ever more complex electronic systems,
for example charge and energy transfer in photosynthesis.
However, most electronic structure packages are only equipped to work within the eigenstate formulation.
There are many unanswered questions about how electron correlation affects the salient features of electronic dynamics.
Furthermore, relative to vibrational dynamics, analysis is complicated by the indistinguishability of those electrons that
move from those electrons that do not.
Our work in this area addresses the dual challenges of simulating correlated many-electron dynamics and interpreting
the results.
Project publications. Click sidebar to see all publications.
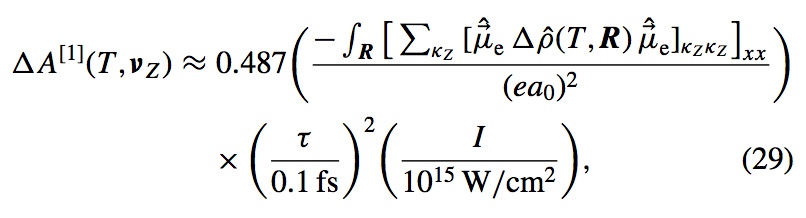
|
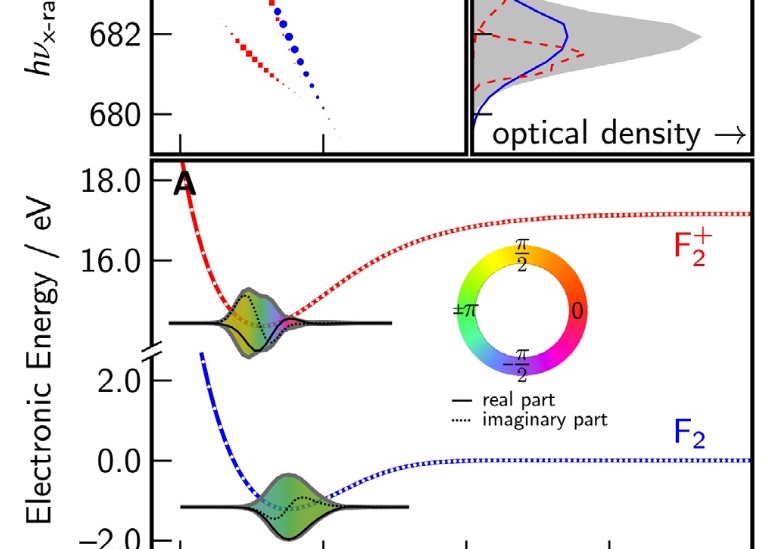
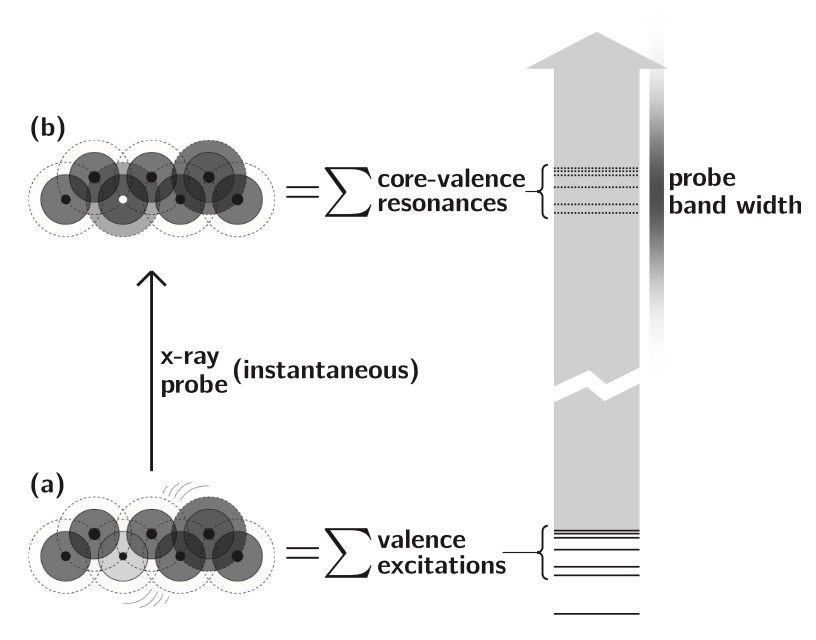
Anticipating the experimental realization of attosecond pulses with photon energies of a few hundred eV to 1 keV, we have developed a formalism that connects the evolution of a UV-pumped nonstationary electronic state to an x-ray probe signal, using the one-electron reduced density operator. The electronic states we wish to follow evolve on time scales of a few femtoseconds, and the valence occupancy structure of these states can be probed, resolved in both space and time, by taking advantage of the inherent locality of core–valence transitions and the comparatively short time scale on which they can be produced. The time-dependent reduced density operator is an intuitively simple quantity, representing the dynamic occupancy structure of the valence levels, but it is well defined for an arbitrary many-body state. This article outlines the connection between the complexities of many-body theory and an intuitive picture of dynamic local orbital occupancy.
|
|
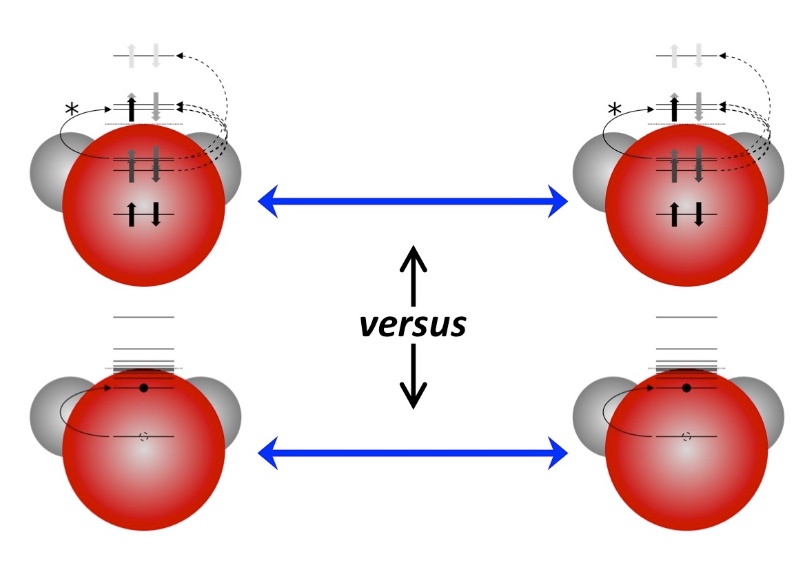
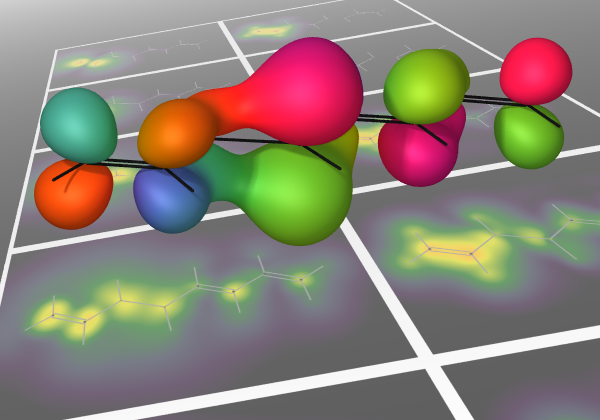
As experimental techniques begin to probe electronic motions in increasing detail, the need is arising for compact and informative visualisations of simulations of such processes. The inherent challenge is that a full many-electron wavefunction is a high-dimensional object, representing the complicated correlations of strongly repulsive bodies in a small molecular volume. A general procedure is needed to distill this to a smaller amount of information that does not rely on any specific level of approximation. The result should allow for easy and intuitive interpretation while drawing out nontrivial aspects of the underlying many-body dynamics, such as the complex phase information inherent of a nonstationary state. Current visualisation schemes based on physical observables or the qualitative information contained in simple wavefunctions, such as time-dependent configuration-interaction-singles (TD-CIS) and time-dependent self-consistent-field (TD-SCF), are discussed. This information is compared to an analysis based on the one-body reduced density operator (1-RDO), which is well-defined for general wavefunctions. It is seen that the distinction between two paradigms of many-body dynamics, electron transport and energy transport, is reflected in the coherences of a difference-1-RDO, or lack thereof.
|
Invited article, author profile: pg. 12 of same issue.
 Also the cover article Also the cover article
 An image from this article was featured as the "back scatter" in Physics Today, Phys. Today 67-6 68 (2014). An image from this article was featured as the "back scatter" in Physics Today, Phys. Today 67-6 68 (2014).
|
|
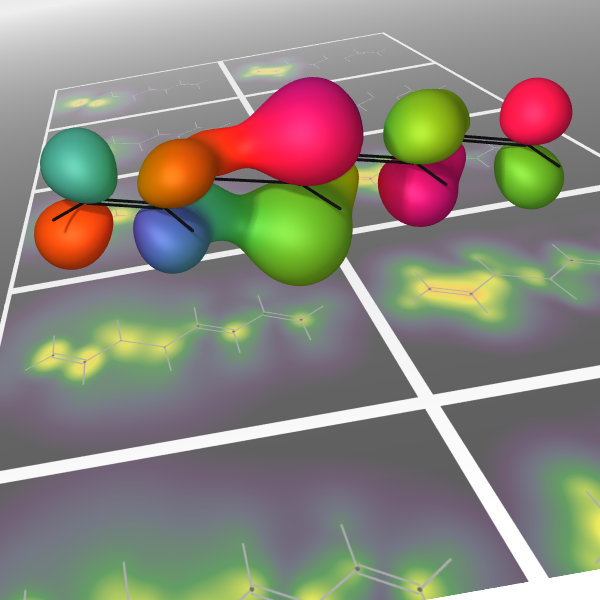
Numerical experiments are performed using a recently published formalism [Phys. Rev. A 88, 013419 (2013)] for computing the transient absorption of an attosecond x-ray probe by a molecule in a nonstationary valence- excited state. This study makes use of an all-electron correlated electronic-structure model to compute electronic dynamics ensuing after a localized excitation on a model chromophore group. Simulated absorption of a delayed attosecond pulse is then used to investigate the presence of an extra valence electron or vacancy around atoms of a given element as a function of time, by tuning the carrier frequency to the associated core-valence energy gap. We show correlations between the predicted absorption of such pulses and visualizations of the particle and hole locations in test molecules. Given the strong role played by the relative orientation of the molecules and the probe polarization, results are presented for a few different alignment schemes. For the molecules studied, effective pump-induced alignment is sufficient to recover easily interpreted information.
|
|
 go to UOP homepage
go to UOP homepage
 © Copyright 2012–2026
© Copyright 2012–2026