As photon energy increases, so does the available bandwidth.
This makes possible extremely short pulses, from just a few femtoseconds down to tens of attoseconds.
This is sufficient resolution to probe the fastest vibrations and even typical electronic motions in real time.
This technology is presently being developed only in the most advanced laboratories around the world,
but as expertise grows and spreads, it will reface our understanding of chemical
and electronic dynamics.
Such studies are in need of new theoretical tools, since existing quantum chemistry packages
focus mostly on the ground state and a handful of low-lying excited states, well below the dense manifold of states at x-ray energies.
We are developing the theoretical framework and associated computational tools to support these advanced experimental explorations.
Project publications. Click sidebar to see all publications.
|
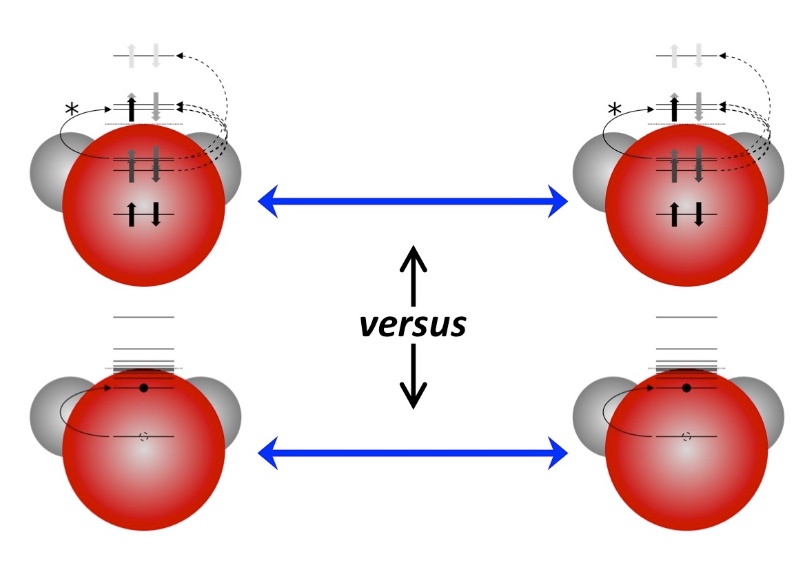
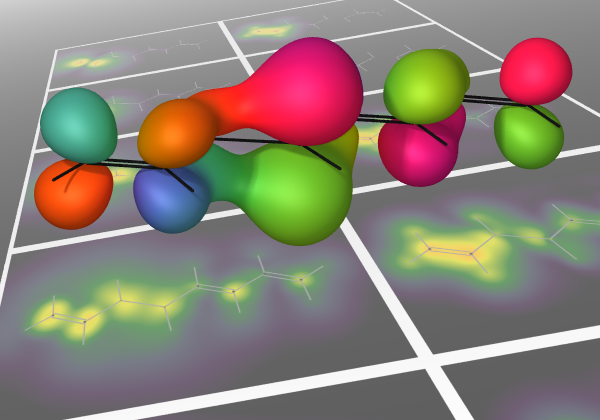
Anticipating the experimental realization of attosecond pulses with photon energies of a few hundred eV to 1 keV, we have developed a formalism that connects the evolution of a UV-pumped nonstationary electronic state to an x-ray probe signal, using the one-electron reduced density operator. The electronic states we wish to follow evolve on time scales of a few femtoseconds, and the valence occupancy structure of these states can be probed, resolved in both space and time, by taking advantage of the inherent locality of core–valence transitions and the comparatively short time scale on which they can be produced. The time-dependent reduced density operator is an intuitively simple quantity, representing the dynamic occupancy structure of the valence levels, but it is well defined for an arbitrary many-body state. This article outlines the connection between the complexities of many-body theory and an intuitive picture of dynamic local orbital occupancy.
|
|
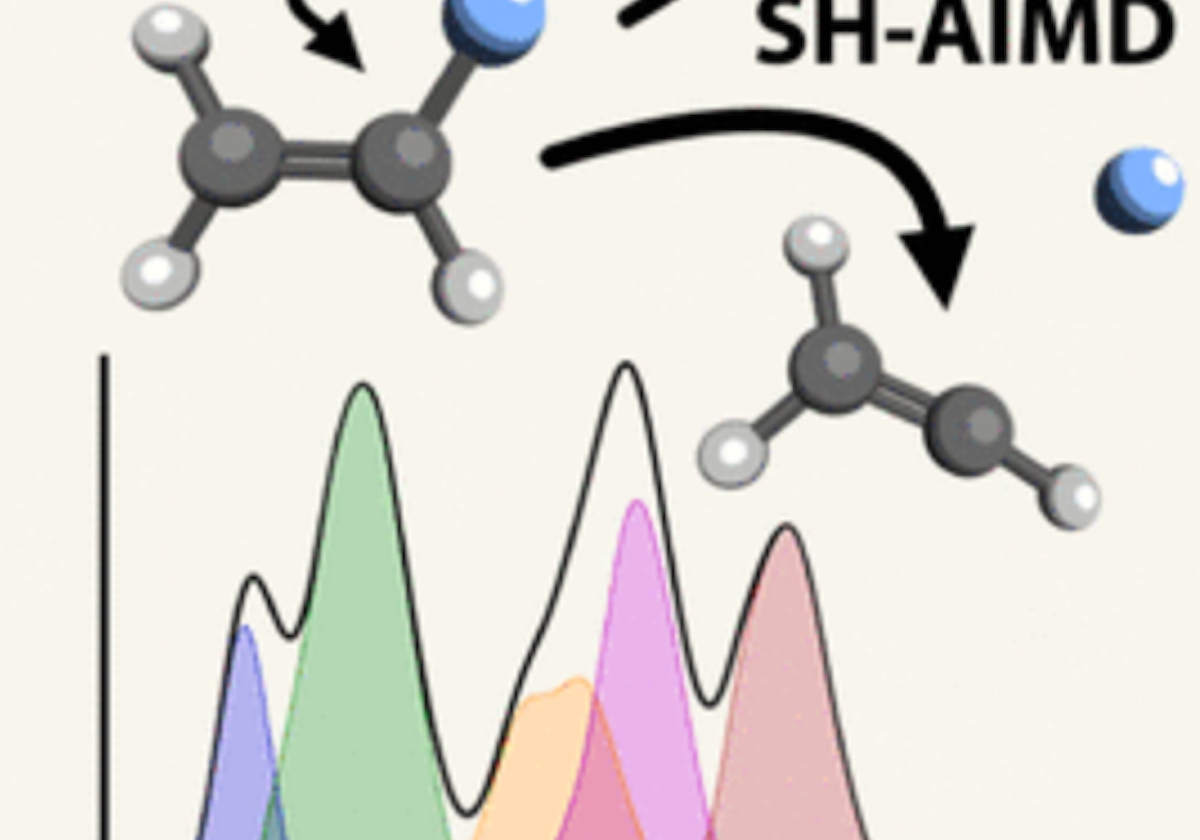
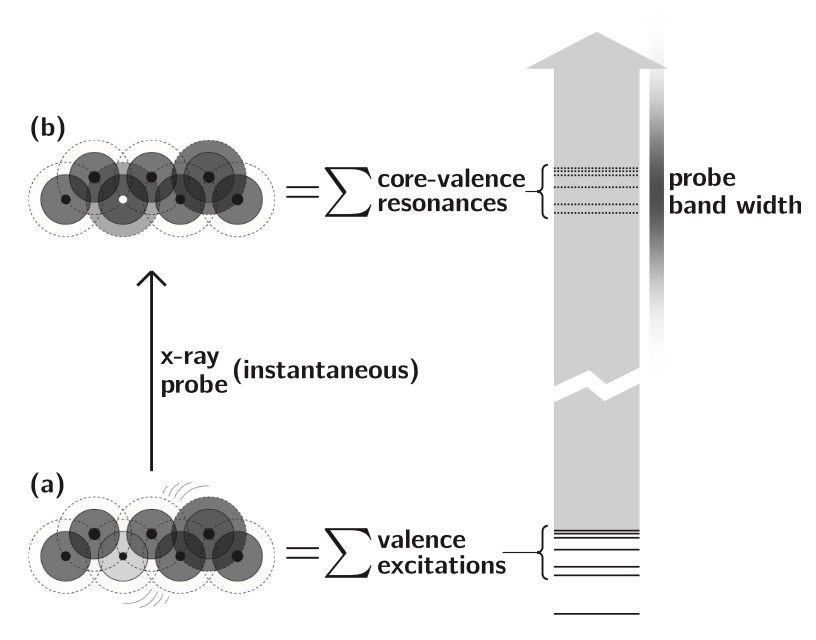
Numerical experiments are performed using a recently published formalism [Phys. Rev. A 88, 013419 (2013)] for computing the transient absorption of an attosecond x-ray probe by a molecule in a nonstationary valence- excited state. This study makes use of an all-electron correlated electronic-structure model to compute electronic dynamics ensuing after a localized excitation on a model chromophore group. Simulated absorption of a delayed attosecond pulse is then used to investigate the presence of an extra valence electron or vacancy around atoms of a given element as a function of time, by tuning the carrier frequency to the associated core-valence energy gap. We show correlations between the predicted absorption of such pulses and visualizations of the particle and hole locations in test molecules. Given the strong role played by the relative orientation of the molecules and the probe polarization, results are presented for a few different alignment schemes. For the molecules studied, effective pump-induced alignment is sufficient to recover easily interpreted information.
|
|
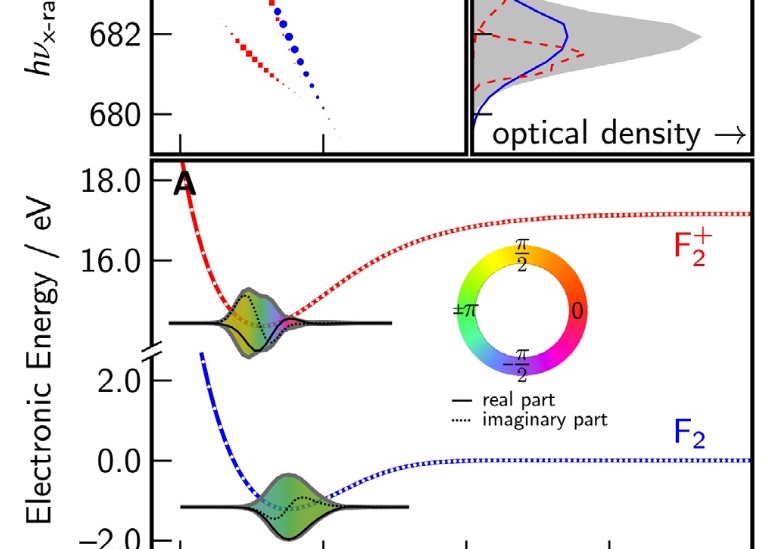
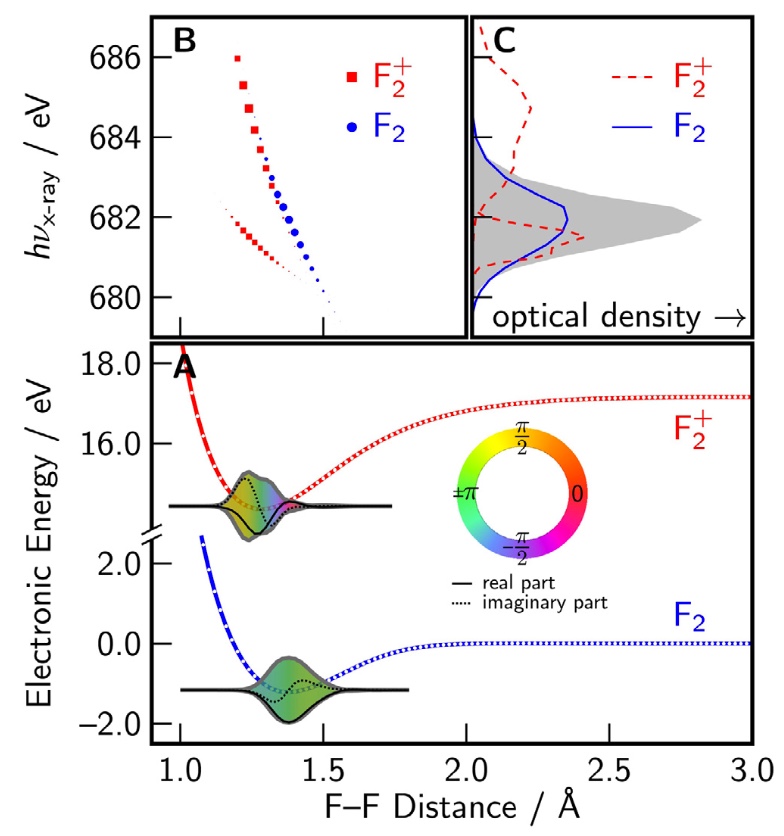
Femtosecond and attosecond X-ray transient absorption experiments are becoming increasingly sophisticated tools for probing nuclear dynamics. In this work, we explore and develop theoretical tools needed for interpretation of such spectra, in order to characterize the vibrational coherences that result from ionizing a molecule in a strong IR field. Ab initio data for F2 is combined with simulations of nuclear dynamics, in order to simulate time-resolved X-ray absorption spectra for vibrational wavepackets after coherent ionization at 0 K and at finite temperature. Dihalogens pose rather difficult electronic structure problems, and the issues encountered in this work will be reflective of those encountered with any core–valence excitation simulation when a bond is breaking. The simulations reveal a strong dependence of the X-ray absorption maximum on the locations of the vibrational wave packets. A Fourier transform of the simulated signal shows features at the overtone frequencies of both the neutral and the cation, which reflect spatial interferences of the vibrational eigenstates. This provides a direct path for implementing ultrafast X-ray spectroscopic methods to visualize coherent nuclear dynamics.
|
|
 go to UOP homepage
go to UOP homepage
 © Copyright 2012–2026
© Copyright 2012–2026