|
|
|
|
|
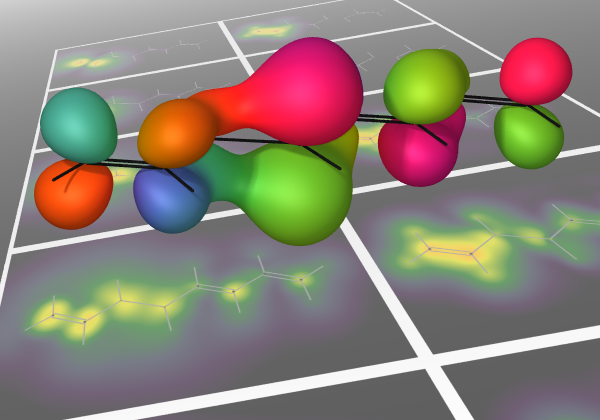
Excitonic
Coupled Cluster
|
Impulsive
Dissociation
|
X-ray Photoelectron
Spectra
|
Time-resolved
X-ray Spectroscopy
|
Ultrafast
Electron Dynamics
|
Click on an image to see a project description.
We are dedicated to a fundamental understanding of electron correlation and its effects on chemistry.
Projects include developing high-accuracy wavefunction-based methods for large systems,
performing ab initio molecular dynamics of open-shell systems,
studying the x-ray spectroscopy of solids (static) and isolated molecules (time-resolved),
and investigating the fundamentals of correlated electronic dynamics.
All of these projects should culminate in better understanding of chemical reactions,
including energy and electron transfer, particularly in the condensed phase and at interfaces.
One of the major shortcomings of modern fragment-based electronic structure methods is that electron correlation (if included)
is handled at the level of individual electrons.
This project consists of two theoretical pieces.
First, the Hamiltonian for a supersystem is rigorously renormalized to be in terms of fluctuations of entire subsystems
(among internally correlated electronic states).
Second, familiar electronic structure methods, notably coupled-cluster theory, can be applied to this
renormalized Hamiltonian.
Initial tests of accuracy per cost are quite promising, and the present bottleneck is the building of the effective Hamiltonian.
Major areas of work consist of defining reusable fragment states that describe well their interactions within a variety of contexts
and representing information about these states efficiently.
The steps beyond are to apply this model to systems of wider interest and implement the theoretically straightforward
generalization to excited states.
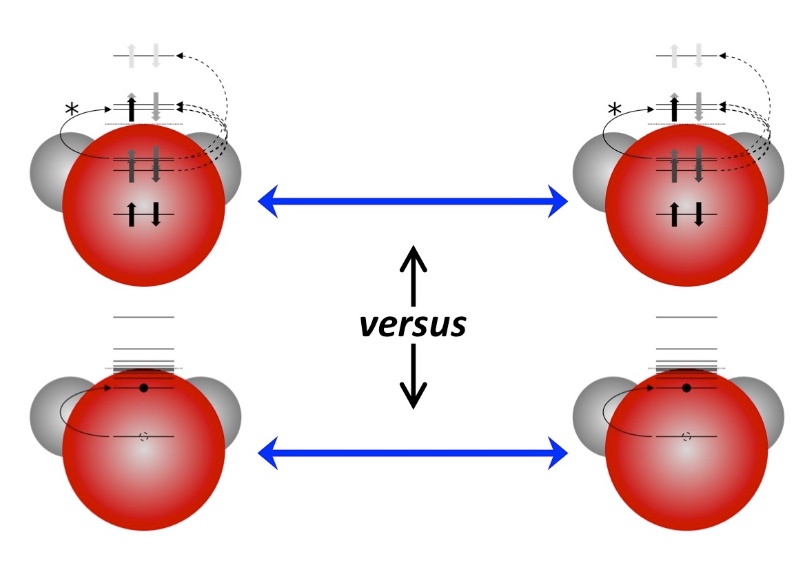
A couple of elementary open problems in the electronic structure theory of solids
are the sources of extra background electrons in x-ray photoelectron spectroscopy
and the asymmetric shapes of some peaks.
We have made breakthroughs on understanding the mechanisms of both phenomena.
In the case of the background contribution, a many-body effect was proposed, by which
intensity borrowing from a deeper core polarization increases the total number of liberated electron.
This hypothesis is supported well by the experiments of our collaborators.
In addition coupled resonances have been proposed to explain peak asymmetries.
This hypothesis has been supported numerically and is likely due to d-shell rearrangments on a core-hole site.
We are now working on an approach to semi-quantitative (trend-predictive) simulation and understanding.
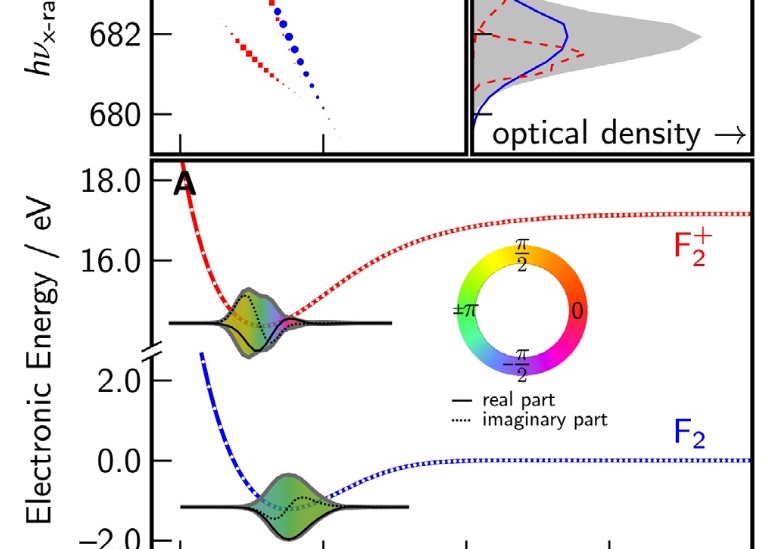
As photon energy increases, so does the available bandwidth.
This makes possible extremely short pulses, from just a few femtoseconds down to tens of attoseconds.
This is sufficient resolution to probe the fastest vibrations and even typical electronic motions in real time.
This technology is presently being developed only in the most advanced laboratories around the world,
but as expertise grows and spreads, it will reface our understanding of chemical
and electronic dynamics.
Such studies are in need of new theoretical tools, since existing quantum chemistry packages
focus mostly on the ground state and a handful of low-lying excited states, well below the dense manifold of states at x-ray energies.
We are developing the theoretical framework and associated computational tools to support these advanced experimental explorations.
Real-time quantum dynamics studies are already a mainstay for nuclear motions of complex systems.
Dynamic studies are necessary because the density of states at relevant energies
renders the eigenstate formulation computationally intractable.
We should not expect the situation to be any different as we consider ever more complex electronic systems,
for example charge and energy transfer in photosynthesis.
However, most electronic structure packages are only equipped to work within the eigenstate formulation.
There are many unanswered questions about how electron correlation affects the salient features of electronic dynamics.
Furthermore, relative to vibrational dynamics, analysis is complicated by the indistinguishability of those electrons that
move from those electrons that do not.
Our work in this area addresses the dual challenges of simulating correlated many-electron dynamics and interpreting
the results.
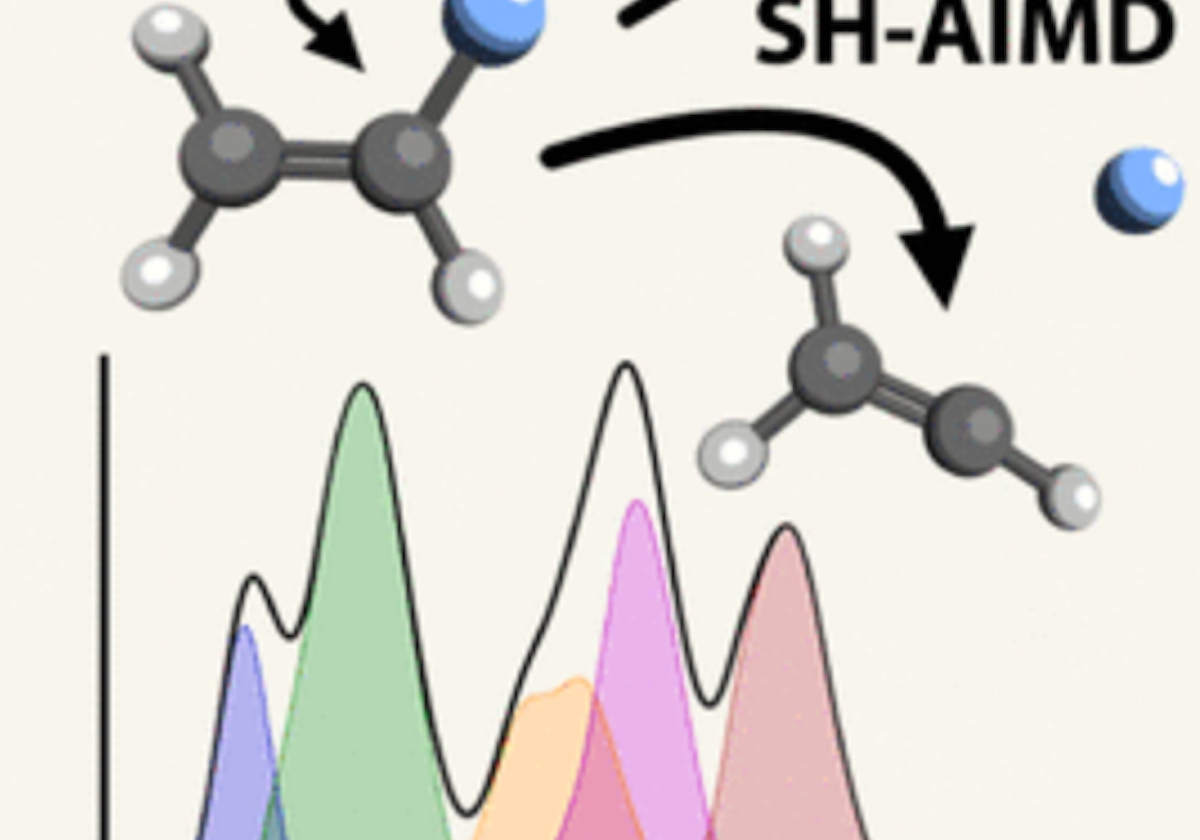
The common understanding of post-ionization fragmentation of molecules is that any excess
electronic energy left in the molecule is quickly converted to vibrational energy, which
then dissociates along different coordinates in a purely statistical manner,
controlled by transition-state theory.
Although the possibilities of other mechanisms have been acknowleged (roaming, impulsive
dissociation), these are computationally demanding to simulate.
In particular, the electronic states of these open-shell species are complex, especially
as bonds also break.
In our work, this is compounded when a dissociation furthermore takes place on
an excited-state surface.
We have done a mixture of intense computational work and reductive theoretical distillation
to understand impulsive, excited-state dissociations of molecular ions.
| See publications 30 and 34. | [Back to overview] |

Dr. Anthony D. Dutoi, Associate Professor of Chemistry
I grew up in Jefferson City, Missouri, and studied chemistry at Saint Louis University, working with Ronald See and graduating with highest honors.
I undertook a year of independent study as a Fulbright Scholar in Göttingen, taking on a research project in vibrational spectroscopy with Peter Botschwina.
I obtained my PhD under the supervision of Martin Head-Gordon at the University of California, Berkeley, researching novel approaches to solving the electronic Schrödinger equation for molecules.
I took a postdoctoral position with Tamar Seideman at Northwestern University, looking at the electron dynamics underlying high-order harmonic generation.
I then moved to Heidelberg, Germany, with a Humboldt Fellowship to research correlated electron dynamics with Lorenz Cederbaum.
Since 2012, I have been a professor at the University of the Pacific.
|
 go to UOP homepage
go to UOP homepage
 © Copyright 2012–2026
© Copyright 2012–2026