All publications. Click above to see publications for a given project.
|
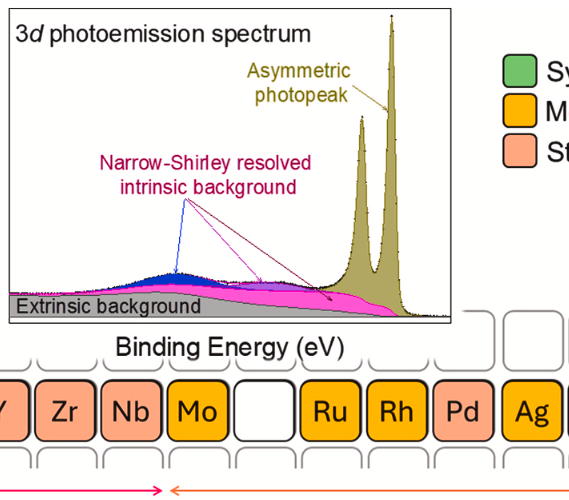
We present a unified, state-of-the-art Coupled-Resonance (CR) and Narrow-Shirley (NS) framework for the comprehensive analysis of 3d core-level photoemission spectra (XPS) of the fifth-row elements (Rb–Te). This novel methodology successfully overcomes the inherent challenges of these spectra, including pronounced main-peak asymmetry and complex, structured backgrounds, to deliver physically meaningful insights. By modeling interference among accessible final-state configurations, CR Type II and Type III lineshapes accurately reproduce asymmetric peak profiles, capturing both the sharp low-binding-energy onsets and gradual high-binding-energy decay. This systematic analysis reveals periodic trends in resonance energies, lifetime widths, and complex intensities that can be compared with first-principles calculations. Concurrently, the NS method resolves the intrinsic background, which is commonly approximated as a monotonic step, to reveal distinct, structured features. These intrinsic background peaks display a novel qualitative correspondence with features in K- and L3-edge Near-Edge X-ray Absorption Fine Structure (NEXAFS). This correlation supports the Interchannel-Coupling with Valence Band Losses (ICVBL) mechanism, involving valence electron excitation into specific unoccupied states. By rigorously isolating extrinsic scattering via Tougaard backgrounds, we ensure a robust distinction between intrinsic excitations and inelastic scattering. In summary, the CR–NS framework is a transferable methodology for resolving complex core-level lineshapes and intrinsic background structure. Although demonstrated here for pure metallic systems, the underlying analytical workflow provides a physically constrained basis for future analysis of more complex compounds and alloys, where separating intrinsic, extrinsic, and chemical-state contributions is essential for reliable electronic-structure interpretation.
|
|
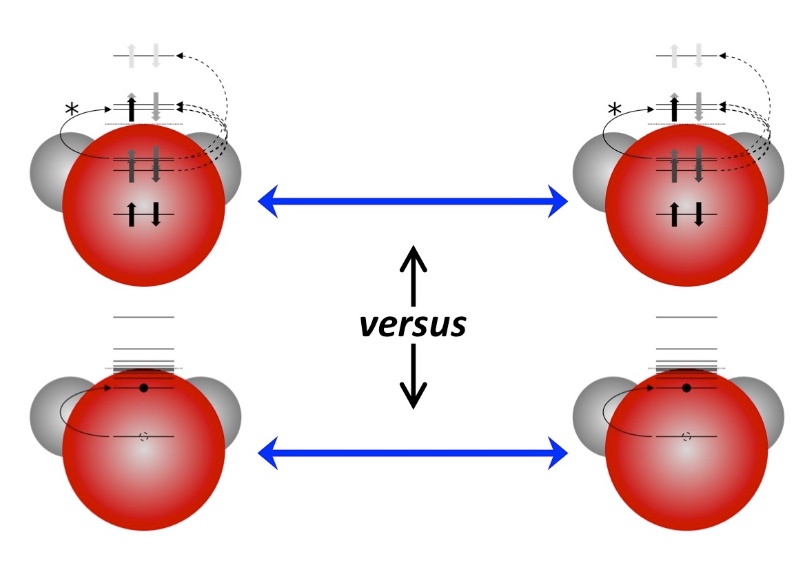
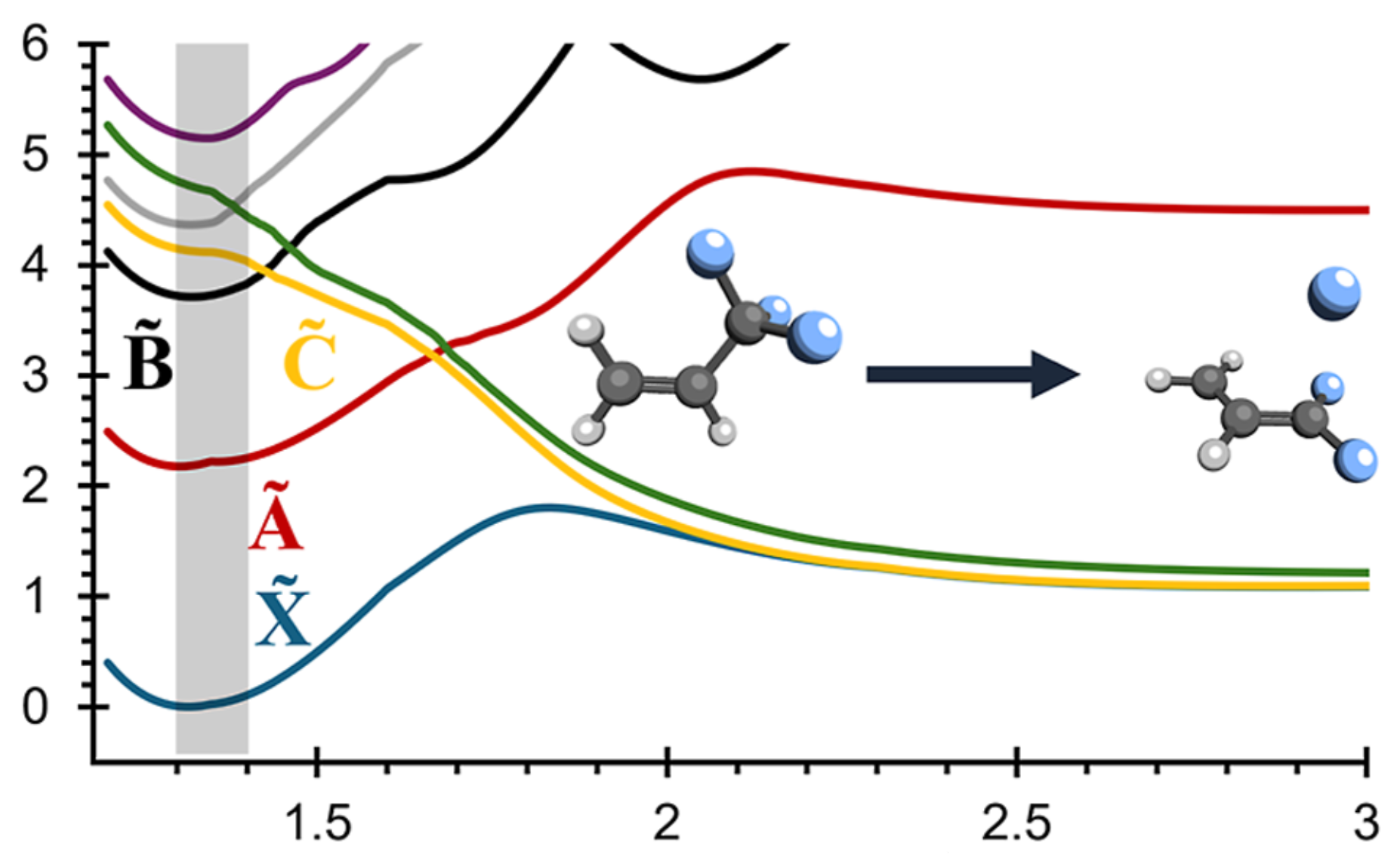
Valence photoionization generates photoions with excess internal energy, often resulting in statistical dissociation, well described by RRKM theory. Yet, numerous small photoions with electronegative substituents (e.g., F, Cl, Br, OH) have been shown to exhibit nonstatistical behavior, undergoing direct dissociation from repulsive electronic states to yield radicals such as F• or OH•, along with their corresponding fragment ions. Here, we present a general, predictive model that rationalizes this mechanism and extends it to molecules bearing electronegative substituents capable of forming 2P (F•, Cl•, Br•) or 2Π (OH•, SH•, N3•, NCO•) radicals. Nonstatistical dissociation arises from ionization of p- or π-localized orbitals on electronegative atoms, producing radical-like fragments unbound to the cationic core. The outcome is governed by three factors: excitation energy, the bonding character of the ionized orbital, and electronic degeneracy between states upon dissociation. Using these criteria, we define four classes of potential energy surfaces, enabling predictive classification of dissociative behavior. Extensive computations on over 50 molecules confirm the model's accuracy and generality.
|
|
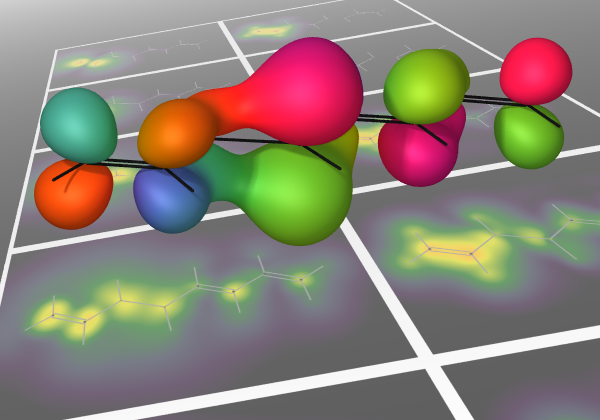
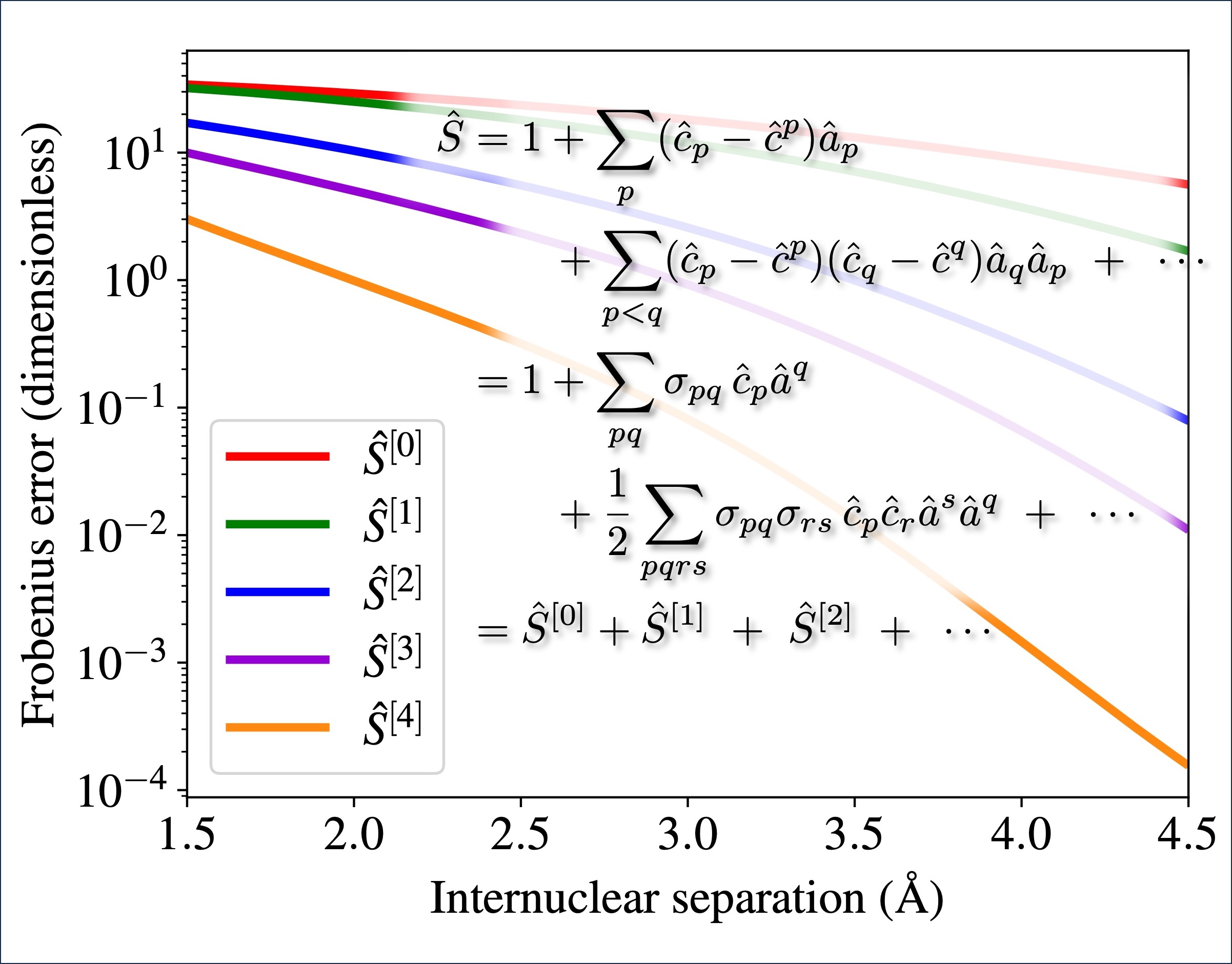
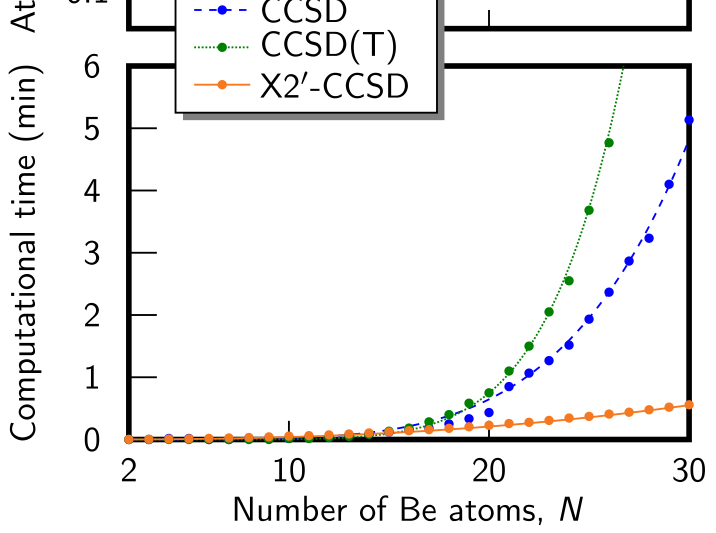
The recently proposed excitonic renormalization framework presents an alternative ansatz to the electronic structure theory of weakly interacting fragments. It makes use of absolutely localized orbitals and correlated states evaluated on isolated fragments, which are then used to recover the interaction in an ab initio manner based on a biorthogonal framework. The correlated monomer information can be heavily truncated, and the Hamiltonian can be expanded in a rapidly converging series, allowing the Hamiltonian to be built and diagonalized in a scalable fashion. However, the methodology still lacks an efficient bottom-up procedure, capable of producing optimized model state spaces for the isolated fragments, without ever building the Hamiltonian in the full monomer state spaces. In order to address this issue, this work presents an algorithm utilizing monomer gradients at three different levels as well as an efficient pre-screening of the determinant space, ensuring compact model state spaces and intermediates. Numerical results are presented for the beryllium dimer, showing that the algorithm is indeed capable of building compact model state spaces, yielding results that closely resemble those of the optimal model state spaces. Furthermore, it is shown that model state spaces, optimized at the zeroth order of the Hamiltonian expansion, can also be used to accurately recover first order results, enabling very efficient optimization, as the optimization can be conducted at a lower order than the targeted final level. Hence, the presented solver completes the excitonic renormalization methodology, forming a polynomially scaling framework.
|
|
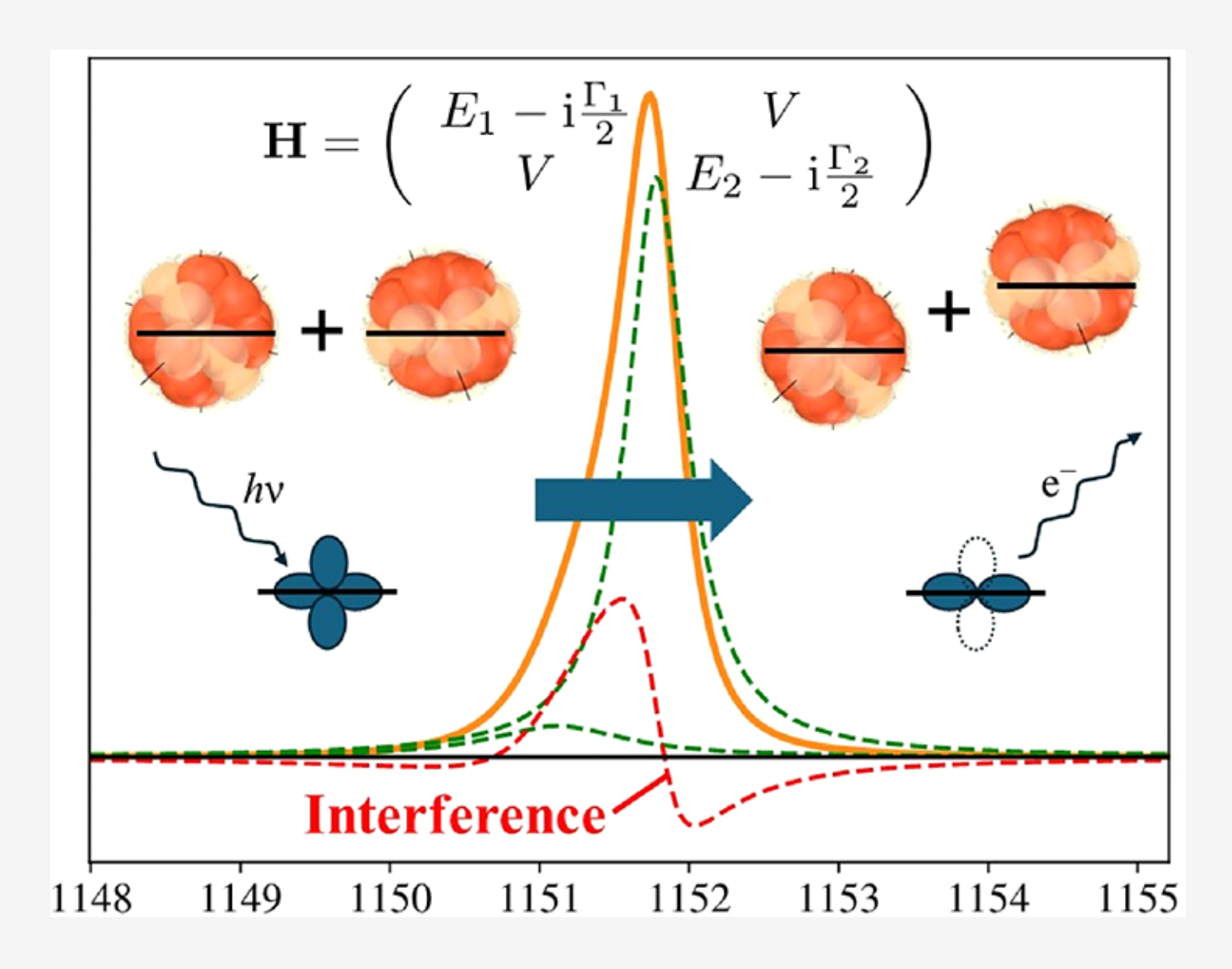
The background signal in X-ray Photoelectron Spectroscopy (XPS) consists of extrinsic and intrinsic components. This study focuses on characterizing the intrinsic background, particularly its structure across an extended binding energy (BE) range beyond the vicinity of the main photoelectron peak. The structure of the intrinsic background in the extended region can be reproduced through a rich set of wide Gaussian peaks. In the near-peak region, the intrinsic background is successfully characterized using the empirical Shirley algorithm. Since the Shirley algorithm fails when applied to extended energy regions by overshooting the experimental signal, a modified version must be employed that decays beyond the near-peak region. We found that a functional form flat near the peak (as in the Shirley algorithm) decays in a Gaussian-like manner for higher binding energies, satisfactorily reproduces the experimental data, and allows for revealing the rest of the rich structure of the intrinsic background. For this reason, we termed it a narrow-Shirley (NS) background. The characterization of the structure of the extended region of the intrinsic background enables the qualitative exploration of its physical origin. Building on previous work linking the near-peak Shirley signal to Interchannel Coupling with Valence Band Losses (ICVBL), we propose and provide qualitative evidence that this ICVBL mechanism is responsible for the entire intrinsic background structure across the extended energy range; this mechanism involves the absorption of a photon by a participating core level. Two of the predictions of the ICVBL mechanism are tested by comparing the structure of the intrinsic background with Auger Electron Spectroscopy (AES) and X-ray Absorption Spectroscopy (XAS) data; a third prediction, the expected modulation of the intrinsic background with photon energies around the threshold of the participating core level, is tested through synchrotron experiments.
|
|
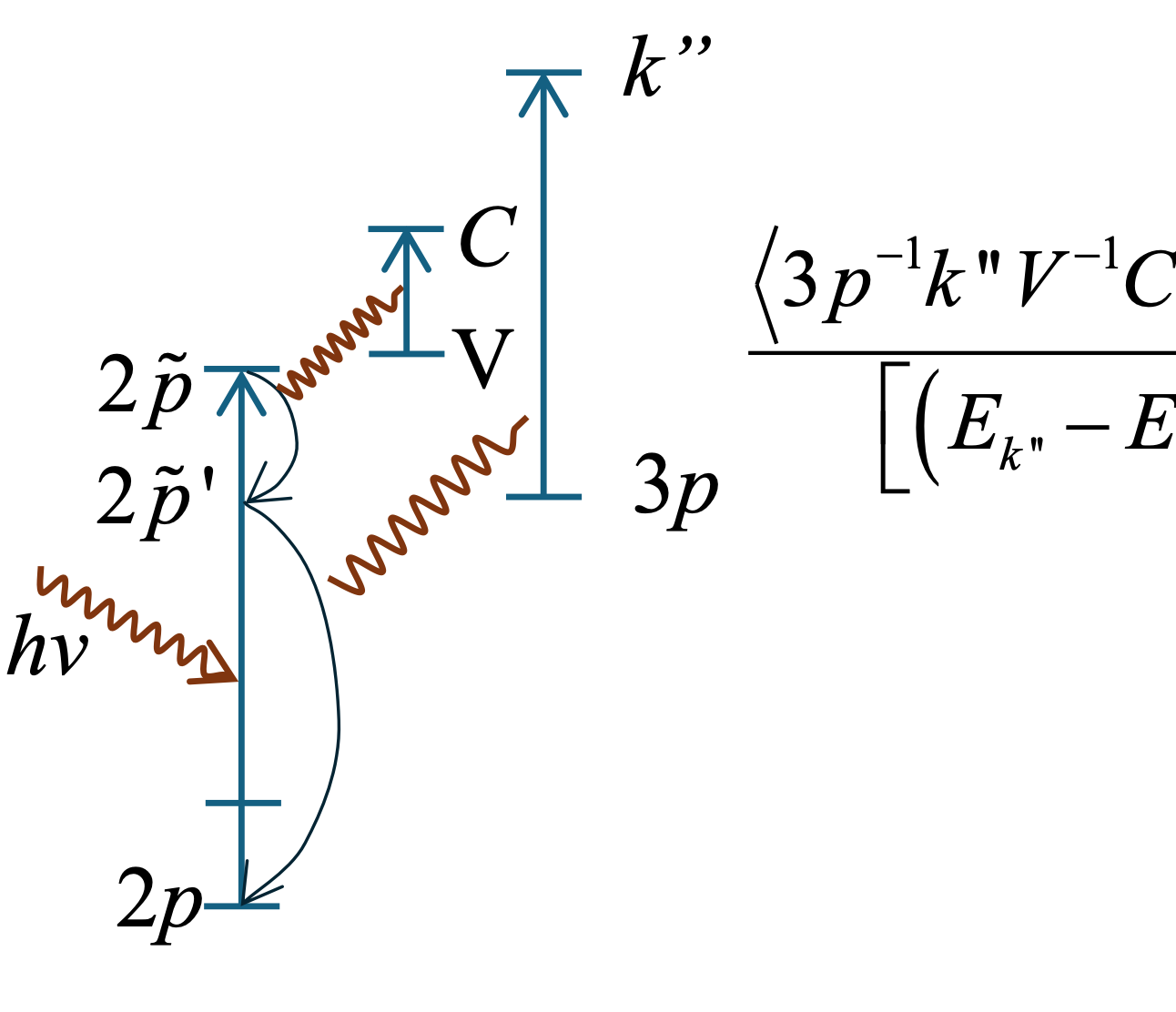
This work introduces the Coupled-Resonances (CR) line shape as a theoretically robust model for analyzing asymmetric peaks in photoelectron spectra, broadly applicable across various materials. The CR line shape extends the conventional Lorentzian distribution by incorporating an interference term that contributes to the peak asymmetry. This new approach addresses limitations of the widely used Doniach-Šunjić (D&S) model, which is often applied beyond its intended scope of metals with high densities of states at the Fermi level due to a lack of viable alternatives. Unlike the DS line shape, the CR model is integrable, enabling its use in precise chemical composition calculations, and it consistently provides superior fits to experimental data. The CR model's versatility is evident in its ability to simplify to a Lorentzian for a single resonance. However, with multiple resonance states, the total line shape is no longer a simple summation of individual peak contributions. Instead, a significant interference term emerges, profoundly contributing to the observed peak asymmetry and shifting the maximum peak intensity. This highlights the critical need to consider interference terms in multiplet calculations of lineshapes. The CR line shape has been implemented in the freely available software, AAnalyzer. While most asymmetric peaks are accurately described by CR Type-II (two resonances), some require CR Type-III (three resonances) for optimal fitting, as demonstrated in the included examples. Ultimately, the CR model offers a more accurate and versatile approach to analyzing asymmetric lineshapes in photoemission spectroscopy, with broad applicability to a wide range of materials, including metals.
|
|
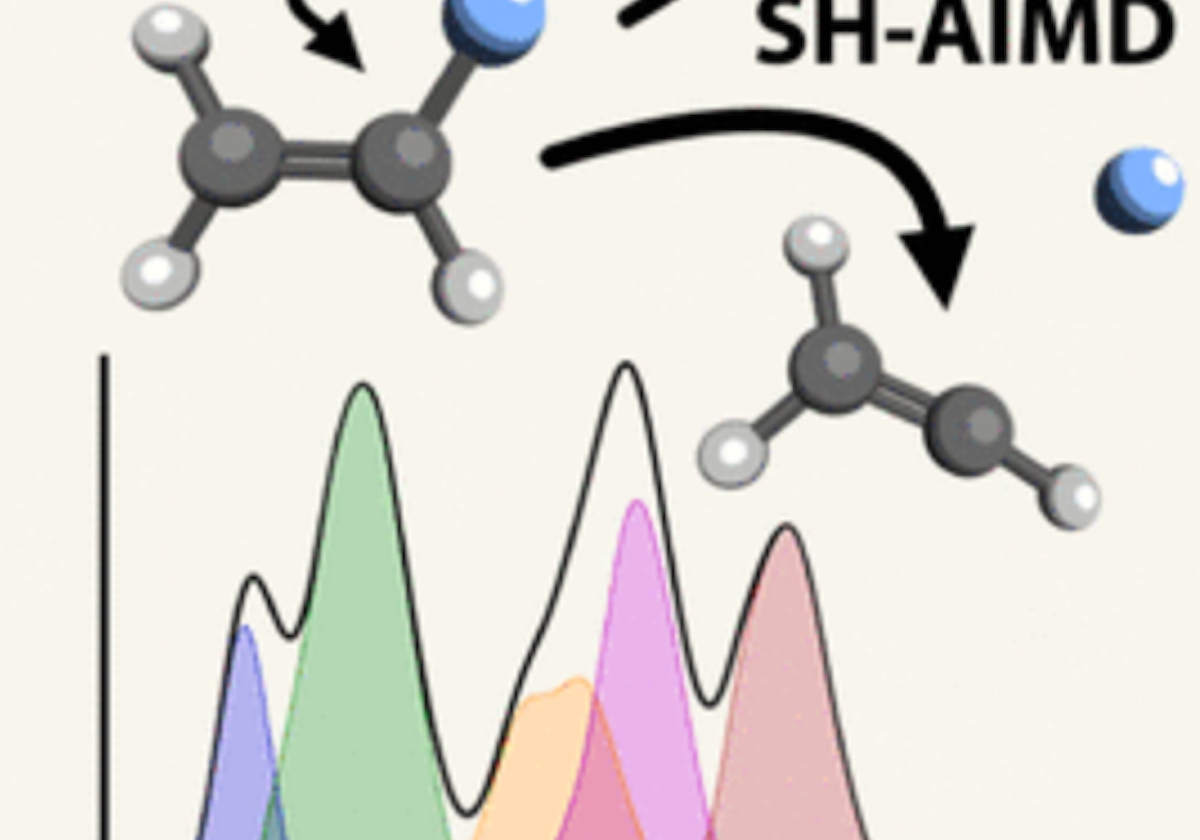
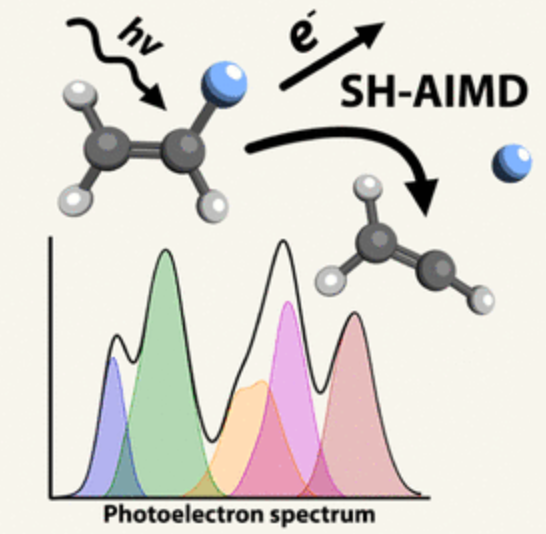
Long-standing debates regarding the dissociative photoionization of vinyl fluoride (fluoroethene) were resolved using large-scale surface-hopping ab initio molecular dynamics (SH-AIMD) simulations. By combining accurate initial condition sampling, electronic cross-section calculations, and SH-AIMD with density functional theory (DFT) and complete active space second-order perturbation theory (CASPT2), we obtained not only qualitative insight into excited-state dynamics but also quantitatively accurate predictions of the photoelectron spectrum, fluorine-loss branching ratios, and translational kinetic energy release distributions for F + C2H3+ products. Statistical dissociation arises from the X̃ 2A″–B̃ 2A′ states, while, in the C̃ 2A″–Ẽ 2A′ states, excited-state dissociation within 50–250 fs dominates. Only CASPT2 captures the formation of an excited triplet C2H3+ fragment, though DFT still reproduces correct branching ratios, as branching pathways are largely determined at short bond distances. Importantly, the previously hypothesized inclusion of autoionizing Rydberg states is not required to match experimental observables.
|
|
Utilizing the sparsity of the electronic structure problem, fragmentation methods have been researched for decades with great success, pushing the limits of ab initio quantum chemistry ever further. Recently, this set of methods has been expanded to include a fundamentally different approach called excitonic renormalization, providing promising initial results. It builds a supersystem Hamiltonian in a second-quantized-like representation from transition-density tensors of isolated fragments, contracted with biorthogonalized molecular integrals. This makes the method fully modular in terms of the quantum chemical methods applied to each fragment and enables massive truncation of the state-space required. Proof-of-principle tests have previously shown that an excitonically renormalized Hamiltonian can efficiently scale to hundreds of fragments, but the ad hoc approach to building the Hamiltonian was not scalable to larger fragments. On the other hand, initial tests of the originally proposed modular Hamiltonian build, presented here, show the accuracy to be poor on account of its non-Hermitian character. In this study, we bridge the gap between these with an operator expansion that is shown to converge rapidly, tending toward a Hermitian Hamiltonian while retaining the modularity, yielding an accurate, scalable method. The accuracy is tested here for a beryllium dimer. At distances near equilibrium and longer, the zeroth-order method is comparable to coupled-cluster singles, doubles, and perturbative triples and the first-order method is comparable to full configuration interaction (FCI). The second-order method agrees with FCI for distances well up the inner repulsive wall of the potential. Deviations occurring at shorter bond distances are discussed along with approaches to scaling to larger fragments.
|
|
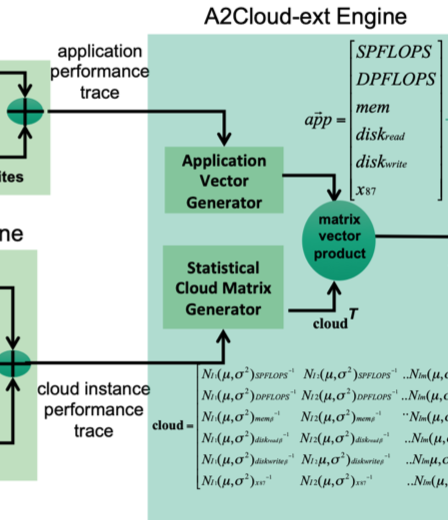
This article proposes a random-forest based A2Cloud framework to match scientific applications with Cloud providers and their instances for high performance. The framework leverages four engines for this task: PERF engine, Cloud trace engine, A2Cloud-ext engine, and the random forest classifier (RFC) engine. The PERF engine profiles the application to obtain performance characteristics, including the number of single-precision (SP) floating-point operations (FLOPs), double-precision (DP) FLOPs, x87 operations, memory accesses, and disk accesses. The Cloud trace engine obtains the corresponding performance characteristics of the selected Cloud instances including: SP floating point operations per second (FLOPS), DP FLOPS, x87 operations per second, memory bandwidth, and disk bandwidth. The A2Cloud-ext engine uses the application and Cloud instance characteristics to generate objective scores that represent the application-to-Cloud match. The RFC engine uses these objective scores to generate two types of random forests to assist users with rapid analysis: application-specific random forests (ARF) and application-class based random forests. The ARF consider only the input application’s characteristics to generate a random forest and provide numerical ratings to the selected Cloud instances. To generate the application-class based random forests, the RFC engine downloads the application profiles and scores of previously tested applications that perform similar to the input application. Using these data, the RFC engine creates a random forest for instance recommendation. We exhaustively test this framework using eight real-world applications across 12 instances from different Cloud providers. Our tests show significant statistical agreement between the instance ratings given by the framework and the ratings obtained via actual Cloud executions.
|
|
A generalized variant of coupled-cluster theory is described here, wherein the degrees of freedom are fluctuations of fragments between internally correlated states. The effects of intra-fragment correlation on the inter-fragment interaction are pre-computed and permanently folded into an effective Hamiltonian, thus avoiding many redundant evaluations of local relaxations associated with coupled fluctuations. A companion article shows that the electronic Hamiltonians of real systems may always be cast in the form required. Two proof-of-principle demonstrations are presented. One uses harmonic oscillators, for which accuracy and algorithm structure can be carefully controlled in comparisons. The other uses small electronic systems to demonstrate compelling accuracy and efficiency, also when inter-fragment electron exchange and charge transfer must be handled. This framework opens a promising path to build finely tunable, systematically improvable methods to capture precise properties of systems interacting with a large number of other systems. Since this scheme is independent of the quality of the states used for the fragments, it can be arbitrarily detailed in terms of the correlation model used for each. The extension to excited states is also straightforward.
|
|
We show here that the Hamiltonian for an electronic system may be written exactly in terms of fluctuation operators that transition constituent fragments between internally correlated states, accounting rigorously for inter-fragment electron exchange and charge transfer. Familiar electronic structure approaches can be applied to the renormalized Hamiltonian. For efficiency, the basis for each fragment can be truncated, removing high-energy local arrangements of electrons from consideration, and effectively defining collective coordinates for the fragments. For a large number of problems, this has the potential to fold the majority of electron correlation into the effective Hamiltonian, and it should provide a robust approach to incorporating difficult electronic structure problems into large systems. The number of terms in the exactly transformed Hamiltonian formally scales quartically with system size, but this can be reduced to quadratic in the mesoscopic regime, to within an arbitrary error tolerance. Finally, all but a linear-scaling number of these terms may be efficiently decomposed in terms of electrostatic interactions between a linear-scaling number of pre-computed transition densities. In a companion article, this formalism is applied to an excitonic variant of coupled-cluster theory.
|
|
The background in X-ray photoelectron spectroscopy data originates, partially, from inelastically scattered photoelectrons. In fact, the current theoretical methods for calculating the background intensity are based on electron energy losses. However, a critical part of the experimental signal, which is known as the Shirley background, cannot be described within the current formalisms. This suggests that the Shirley electrons are not associated with energy losses of photoelectrons and must originate from a different photoexcitation phenomenon with a cross section of its own. We propose a mechanism based on core channeling as the physical origin of the Shirley signal.
|
|
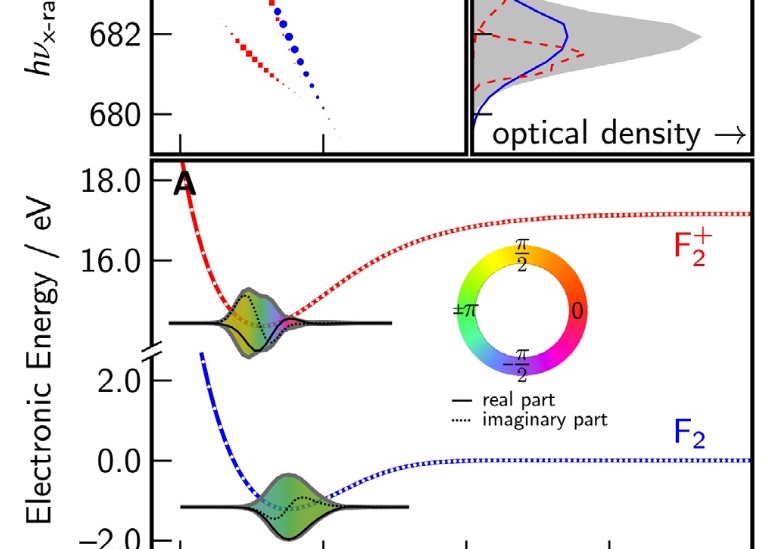
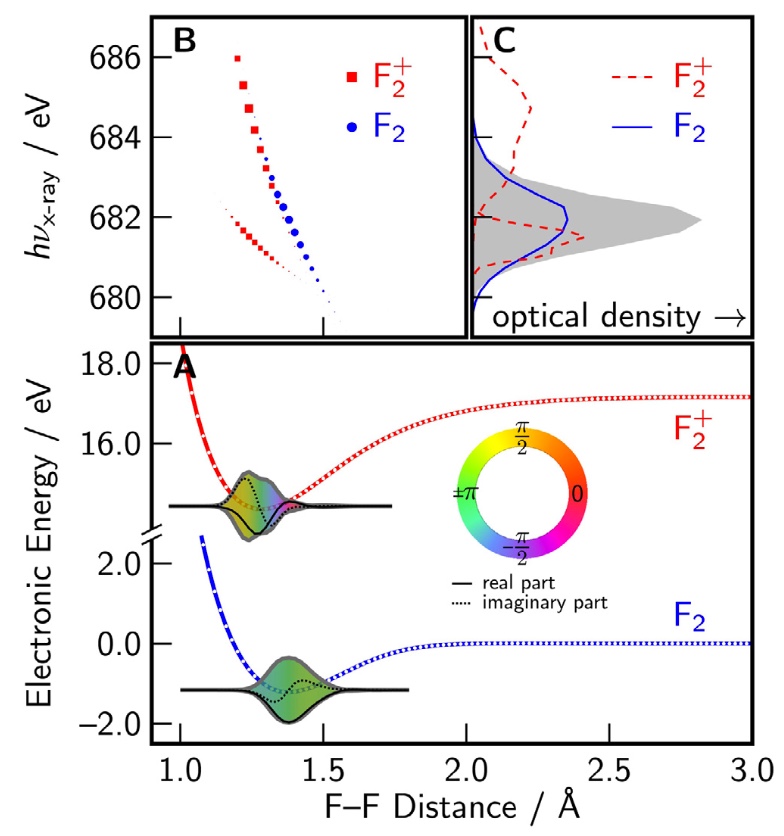
Femtosecond and attosecond X-ray transient absorption experiments are becoming increasingly sophisticated tools for probing nuclear dynamics. In this work, we explore and develop theoretical tools needed for interpretation of such spectra, in order to characterize the vibrational coherences that result from ionizing a molecule in a strong IR field. Ab initio data for F2 is combined with simulations of nuclear dynamics, in order to simulate time-resolved X-ray absorption spectra for vibrational wavepackets after coherent ionization at 0 K and at finite temperature. Dihalogens pose rather difficult electronic structure problems, and the issues encountered in this work will be reflective of those encountered with any core–valence excitation simulation when a bond is breaking. The simulations reveal a strong dependence of the X-ray absorption maximum on the locations of the vibrational wave packets. A Fourier transform of the simulated signal shows features at the overtone frequencies of both the neutral and the cation, which reflect spatial interferences of the vibrational eigenstates. This provides a direct path for implementing ultrafast X-ray spectroscopic methods to visualize coherent nuclear dynamics.
|
23. Advances in Molecular Quantum Chemistry contained in the Q-Chem 4 Program Package
Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr., H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon
[Mol. Phys. 113 184 (2015)]
|

|
A summary of the technical advances that are incorporated in the fourth major release of the Q-Chem quantum chemistry program is provided, covering approximately the last seven years. These include developments in density functional theory methods and algorithms, nuclear magnetic resonance (NMR) property evaluation, coupled cluster and perturbation theories, methods for electronically excited and open-shell species, tools for treating extended environments, algorithms for walking on potential surfaces, analysis tools, energy and electron transfer modelling, parallel computing capabilities, and graphical user interfaces. In addition, a selection of example case studies that illustrate these capabilities is given. These include extensive benchmarks of the comparative accuracy of modern density functionals for bonded and non-bonded interactions, tests of attenuated second order Møller-Plesset (MP2) methods for intermolecular interactions, a variety of parallel performance benchmarks, and tests of the accuracy of implicit solvation models. Some specific chemical examples include calculations on the strongly correlated Cr2 dimer, exploring zeolite-catalysed ethane dehydrogenation, energy decomposition analysis of a charged ter-molecular complex arising from glycerol photoionisation, and natural transition orbitals for a Frenkel exciton state in a nine-unit model of a self-assembling nanotube.
|
|
Numerical experiments are performed using a recently published formalism [Phys. Rev. A 88, 013419 (2013)] for computing the transient absorption of an attosecond x-ray probe by a molecule in a nonstationary valence- excited state. This study makes use of an all-electron correlated electronic-structure model to compute electronic dynamics ensuing after a localized excitation on a model chromophore group. Simulated absorption of a delayed attosecond pulse is then used to investigate the presence of an extra valence electron or vacancy around atoms of a given element as a function of time, by tuning the carrier frequency to the associated core-valence energy gap. We show correlations between the predicted absorption of such pulses and visualizations of the particle and hole locations in test molecules. Given the strong role played by the relative orientation of the molecules and the probe polarization, results are presented for a few different alignment schemes. For the molecules studied, effective pump-induced alignment is sufficient to recover easily interpreted information.
|
|
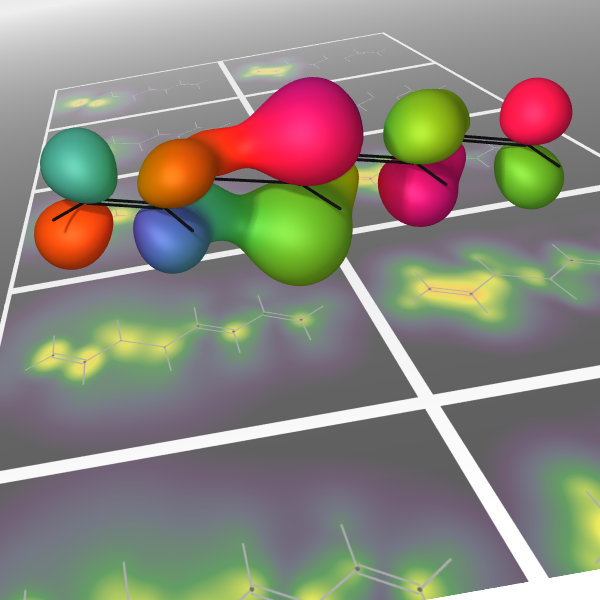
As experimental techniques begin to probe electronic motions in increasing detail, the need is arising for compact and informative visualisations of simulations of such processes. The inherent challenge is that a full many-electron wavefunction is a high-dimensional object, representing the complicated correlations of strongly repulsive bodies in a small molecular volume. A general procedure is needed to distill this to a smaller amount of information that does not rely on any specific level of approximation. The result should allow for easy and intuitive interpretation while drawing out nontrivial aspects of the underlying many-body dynamics, such as the complex phase information inherent of a nonstationary state. Current visualisation schemes based on physical observables or the qualitative information contained in simple wavefunctions, such as time-dependent configuration-interaction-singles (TD-CIS) and time-dependent self-consistent-field (TD-SCF), are discussed. This information is compared to an analysis based on the one-body reduced density operator (1-RDO), which is well-defined for general wavefunctions. It is seen that the distinction between two paradigms of many-body dynamics, electron transport and energy transport, is reflected in the coherences of a difference-1-RDO, or lack thereof.
|
Invited article, author profile: pg. 12 of same issue.
 Also the cover article Also the cover article
 An image from this article was featured as the "back scatter" in Physics Today, Phys. Today 67-6 68 (2014). An image from this article was featured as the "back scatter" in Physics Today, Phys. Today 67-6 68 (2014).
|
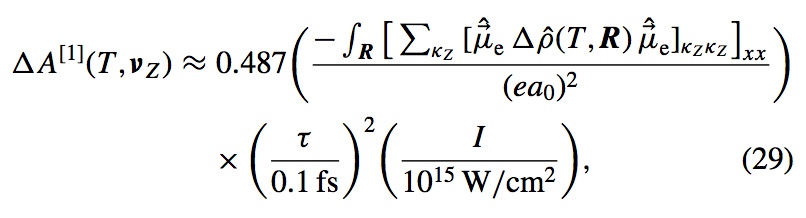
|
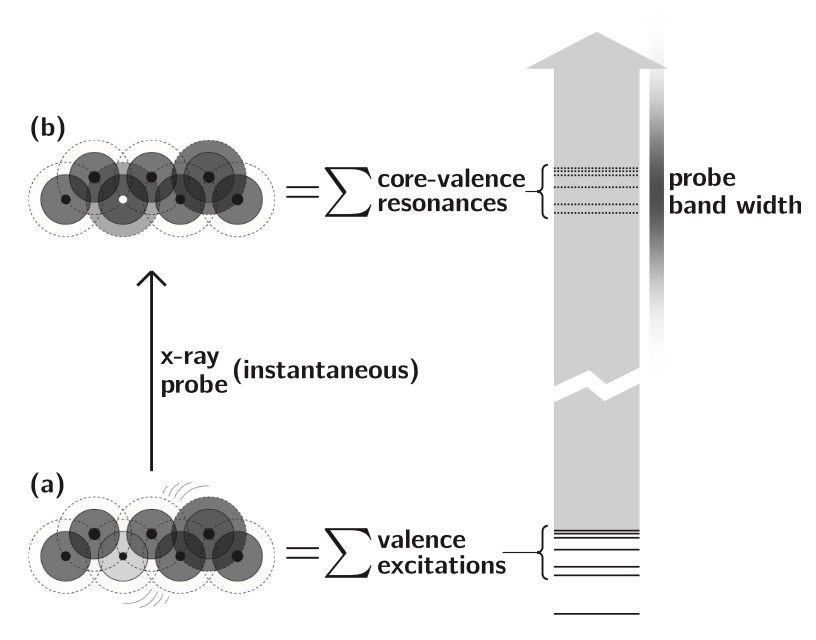
Anticipating the experimental realization of attosecond pulses with photon energies of a few hundred eV to 1 keV, we have developed a formalism that connects the evolution of a UV-pumped nonstationary electronic state to an x-ray probe signal, using the one-electron reduced density operator. The electronic states we wish to follow evolve on time scales of a few femtoseconds, and the valence occupancy structure of these states can be probed, resolved in both space and time, by taking advantage of the inherent locality of core–valence transitions and the comparatively short time scale on which they can be produced. The time-dependent reduced density operator is an intuitively simple quantity, representing the dynamic occupancy structure of the valence levels, but it is well defined for an arbitrary many-body state. This article outlines the connection between the complexities of many-body theory and an intuitive picture of dynamic local orbital occupancy.
|
|
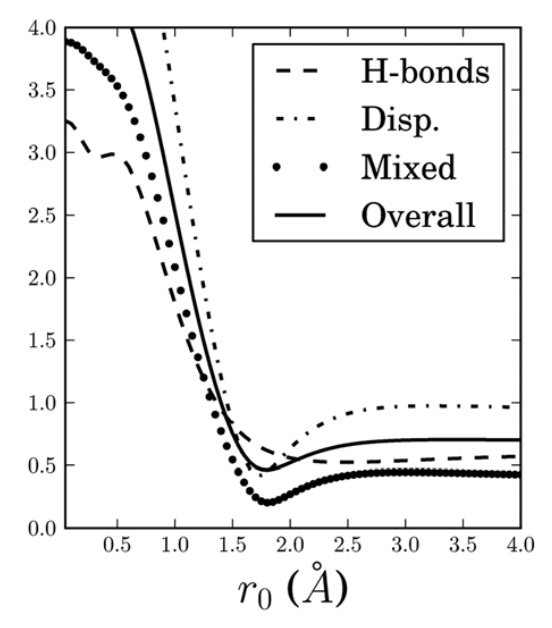
Attenuated second-order Møller-Plesset perturbation theory (MP2) within the finite aug-cc-pVTZ (aTZ) basis set is developed for inter- and intra-molecular non-bonded interactions. A single attenuation parameter is optimized on the S66 database of 66 intermolecular interactions, leading to a very large RMS error reduction by a factor of greater than 5 relative to standard MP2/aTZ. Attenuation introduces an error of opposite sign to basis set superposition error (BSSE) and overestimation of dispersion inter- actions in finite basis MP2. A variety of tests including the S22 set, conformer energies of peptides, alkanes, sugars, sulfate-water clusters, and the coronene dimer establish the transferability of the MP2(terfc, aTZ) model to other inter and intra-molecular interactions. Direct comparisons against attenuation in the smaller aug-cc-pVDZ basis shows that MP2(terfc, aTZ) often significantly outperforms MP2(terfc, aDZ), although at higher computational cost. MP2(terfc, aDZ) and MP2(terfc, aTZ) often outperform MP2 at the complete basis set limit. Comparison of the two attenuated MP2 models against each other and against attenuation using non-augmented basis sets gives insight into the error cancellation responsible for their remarkable success.
|
Publications prior to University of the Pacific
-
A. D. Dutoi and L. S. Cederbaum
"An Excited Electron Avoiding a Positive Charge"
J. Phys. Chem. Lett. 2 2300 (2011)
Highlighted in "News & Views" Nat. Chem. 4 8 (2012)
-
A. D. Dutoi, M. Wormit and L. S. Cederbaum
"Ultrafast Charge Separation Driven by Differential Particle and Hole Mobilities"
J. Chem. Phys. 134 024303 (2011)
Figure from this article reproduced in Nat. Photonics 8 165 (2014)
-
A. D. Dutoi, L. S. Cederbaum, M. Wormit, J. H. Starcke and A. Dreuw
"Tracing Molecular Electronic Excitation Dynamics in Real Time and Space"
J. Chem. Phys. 132 144302 (2010)
-
A. Dreuw, J. Plötner, M. Wormit, M. Head-Gordon and A. D. Dutoi
"An Additive Long-range Potential to Correct for the Charge-transfer Failure of Time-dependent Density Functional Theory"
in Progress in Physical Chemistry ed. F. M. Dolg, v. 3, p. 21 (Oldenbourg, München, 2010)
-
A. Dreuw, J. Plötner, M. Wormit, M. Head-Gordon and A. D. Dutoi
"An Additive Long-range Potential to Correct for the Charge-transfer Failure of Time-dependent Density Functional Theory"
Z. Phys. Chem. 224 311 (2010)
-
S. Ramakrishna, P. A. J. Sherrat, A. D. Dutoi and T. Seideman
"Origin and Implication of Ellipticity in High-order Harmonic Generation from Aligned Molecules"
Phys. Rev. A 81 021802 (2010)
-
J. A. Parkhill, J.-D. Chai, A. D. Dutoi, and M. Head-Gordon
"The Exchange Energy of a Uniform Electron Gas Experiencing a New, Flexible Range Separation"
Chem. Phys. Lett. 478 283 (2009)
-
A. D. Dutoi and M. Head-Gordon
"A Study of the Effect of Attenuation Curvature on Molecular Correlation Energies by Introducing an Explicit Cut-off Radius into Two-electron Integrals"
J. Phys. Chem. A 112 2110 (2008)
-
Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer III, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon
"Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package"
Phys. Chem. Chem. Phys. 8 3172 (2006)
-
A. D. Dutoi and M. Head-Gordon
"Self-interaction Error of Local Density Functionals for Alkali-halide Dissociation"
Chem. Phys. Lett. 422 230 (2006)
-
J. E. Subotnik, A. D. Dutoi and M. Head-Gordon
"Fast, Localized, Orthonormal Virtual Orbitals which Depend Smoothly on Nuclear Coordinates"
J. Chem. Phys. 123 114108 (2005)
-
A. Aspuru-Guzik*, A. D. Dutoi*, P. J. Love and M. Head-Gordon
"Simulated Quantum Computation of Molecular Energies"
Science 309 1704 (2005)
*equal authorship
-
P. E. Maslen, A. D. Dutoi, M. S. Lee, Y. Shao and M. Head-Gordon
"Accurate Local Approximations to the Triples Correlation Energy: Formulation, Implementation and Tests of 5th-order Scaling Models"
Mol. Phys. 103 425 (2005)
-
A. D. Dutoi, Y. Jung and M. Head-Gordon
"An Orbital-based Definition of Radical and Multiradical Character"
J. Phys. Chem. A 108 10270 (2004)
-
Y. Jung, R. C. Lochan, A. D. Dutoi and M. Head-Gordon
"Scaled Opposite-spin Second-order Møller–Plesset Correlation Energy: An Economical Electronic Structure Method"
J. Chem. Phys. 121 9793 (2004)
-
P. Botschwina, T. Dutoi, M. Mladenovic, R. Oswald, S. Schmatz and H. Stoll
"Theoretical Investigations of Proton-bound Cluster Ions"
Faraday Discuss. 118 433 (2001)
-
R. F. See, A. D. Dutoi, K. W. McConnell and R. M. Naylor
"Geometry of Simple Molecules: Nonbonded Interactions and Not-bonding Orbitals Primarily Determine Observed Geometries"
J. Am. Chem. Soc. 123 2839 (2001)
-
R. F. See, A. D. Dutoi, J. C. Fettinger, P. J. Nicastro and J. W. Ziller
"The Crystal Structures of (p-ClPh)3PO and (p-OMePh)3PO, Including an Analysis of the P-O Bond in Triarylphosphine Oxides"
J. Chem. Crystallogr. 28 893 (1998)
|
 go to UOP homepage
go to UOP homepage
 © Copyright 2012–2026
© Copyright 2012–2026