Molecular dynamics of cationic surfactants with small organic counterions
Amphiphilic systems have many interesting features and applications. Simulating these systems, especially at low concentrations around the critical micelle concentration (CMC), poses a number of challenges. Chief among these are the timescales: hundreds of nanoseconds to microseconds are needed to observe complete mesoscopic equilibrium. With conventional all-atom simulations, this is a problem, as even for a system based on 100 cation/anion pairs, the combination of atom count and simulation time stretches the limits of all but large-scale computing resources.
The answer lies in coarse-grained models, specifically the well-established Martini model. In this model, groups of atoms are represented by beads whose properties reflect those of the underlying chemical groups. Four water molecules are represented by one "W" bead. Tetradecyltrimethylammonium (TTA) is represented by five beads: a charged head group and four non-polar carbon tail beads. Benzene-based anions such as benzoate, phthalate, and ortho-aminobenzoate are represented by four beads, some of them newly parametrized.
These changes in scale allow a timestep of 0.03 ps (30 fs) — 15 times a typical all-atom MD timestep of 2 fs. Thus a system of just under 1 million Martini beads running for 1 μs (1,000 ns) yields reasonable wall-clock run times.
3.4mM
We are working with Dr. Steve Bachover of Saint Mary's College. He is studying the experimental behavior of these systems around the CMC with ITC and NMR. We hope to gain predictive insights for the CMC itself — experiments can help us test the parametrization of the surfactants and ions. In addition, we should be able to understand the mesoscopic changes that occur and are reflected in surface tension and calorimetric measurements.
Conventional clustering analyses are somewhat limited in what they may accomplish. From our work simulating ESI processes, we are able to analyze aggregate size and composition in great detail over the course of a simulation. We can use these analyses to fix the CMC, watch micellization changes, and help characterize shape and extent properties including continuous phases. We are able to characterize the CMC through aggregate distribution: pre-CMC more than 50% of the amphiphiles are dissolved monomers. Above the CMC, there is both a minimum in the aggregate distribution, and monomer concentration is less than 50%.
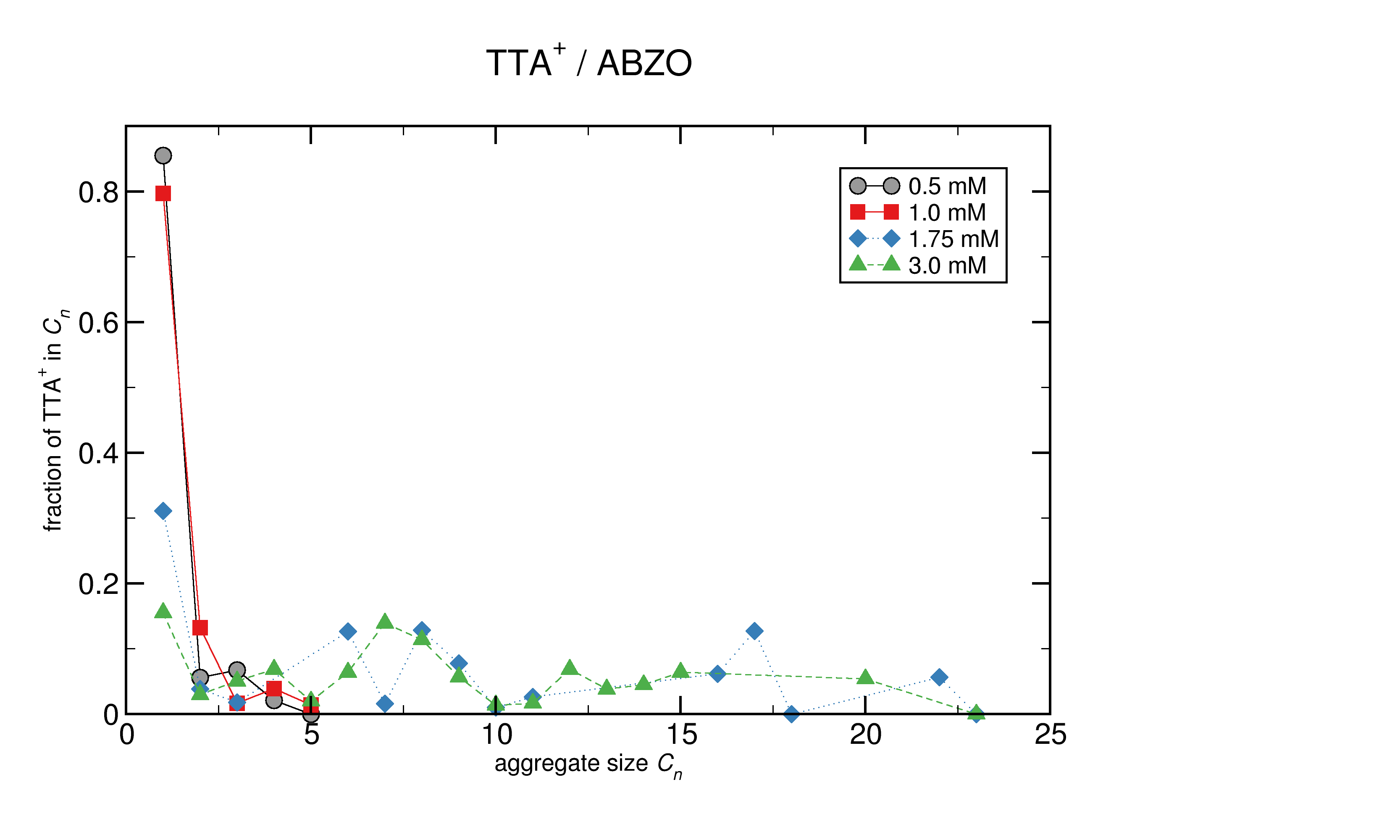
At concentrations near or below the CMC, the anions do not appear to be very involved in micelles. However, different anions do affect the CMC, as observed experimentally, and we do see this in the simulations. At higher concentrations, we expect the anions to be incorporated into the micelles more and more, and certain anions under study could be self-aggregating, apart from the TTA cations.
Questions we'd like to answer:
- What simulation length/time is suitable at different concentrations? It is clear that simulations at low concentration (less than 1.0 mM), below the CMC will equilibrate, or fail to show aggregation in less simulation time. Can this be made predictable?
- Electrostatics are usually handled by the reaction-field method for simulations such as these, as it reduces computational load. How does a more rigorous method such as PME affect the simulation results?
- Martini3 parameterization of new ions, both organic anions and cationic surfactants. This includes checking previous parameterizations that have been done.
- Could we use cluster/aggregation analysis to help characterize more concentrated phases such as wormlike micelles or microemulsion?