CMM 4/10/2026
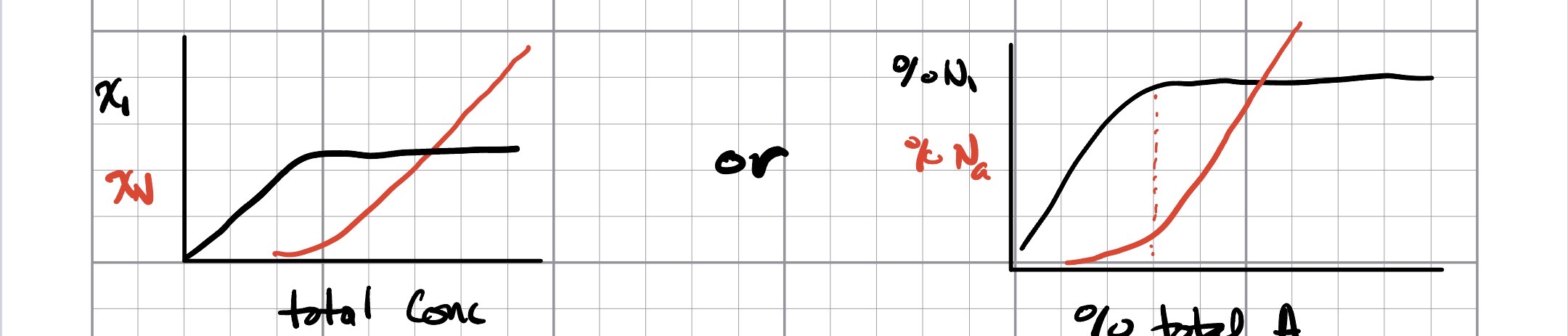
Monomer and aggregate concentration versus solution concentration
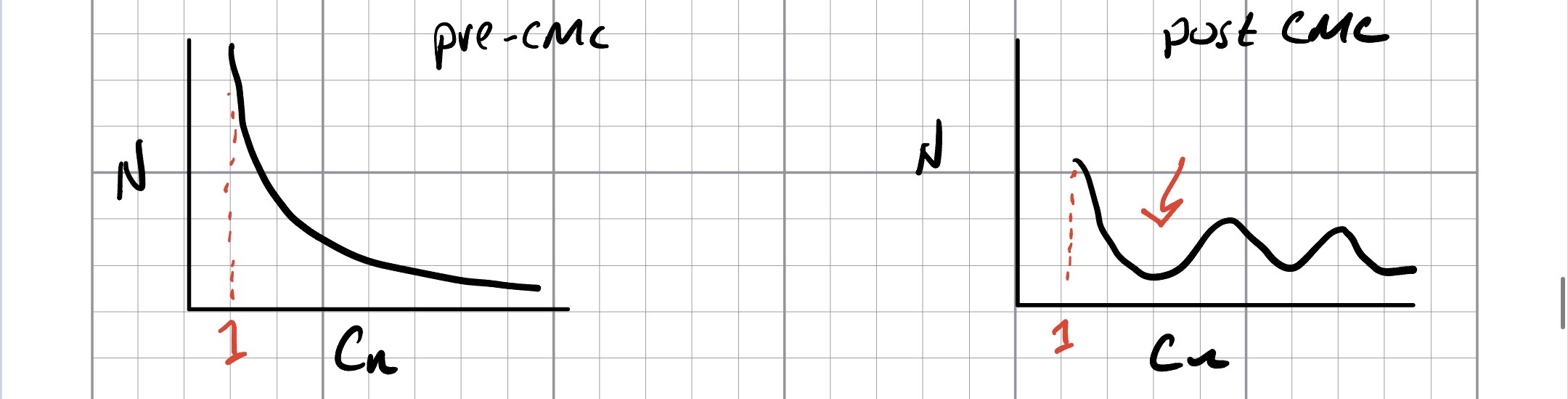
If we can bin the number of amphiphiles in aggregates of size n (Cn) versus n, we should see two behaviors below and above the CMC. Below the CMC, most of the amphiphiles exist as free monomers, so the histogram exponentially (perhaps not literally) decreases from C1 with the fraction close to 1. Above the CMC, there is both a minima in the distribution, and the fraction of monomers is less than 50%:
These things are not directly measurable via experiment, however with simulation we are able to make such an analysis. In the first case, we need to combine measurements from simulations at different concentrations, but with the latter, we can analyze at fixed concentration (per simulation). Both of these analyses will help us fix the CMC according to our model.
It is important to build and follow a consistent file convention. Here is how to do it for this project:
Directory structure: AMPHIPHILE/ANION/CONCENTRATION (that is, each amphiphile will be simulated with a number of anions, and each simulation will be at a different concentration.) For example: TTA/ABZO/1.0mM.
The TTA directory holds all the system martini_v3.0.0 ITP files, along with the TTA ITP and .gro, and water.gro file. The ABZO directory holds the ABZO ITP and .gro files (ABZO_bartender.itp). The simulation directory 1.0mM would have the following files:
In addition, you need the analysis script cmc.tcl; This can live anywhere, but it should live in one place, so you don't end up with many (possibly edited) copies, providing inconsistent data between different simulations. I suggest putting it in the TTA directory. Make sure it is executable:
/bin/bash $ chmod 755 cmc.tcl
This is not simply to visualize your system; removing the water from the system reduces the amount of work the analysis script needs to do. You should at least use VMD to look at the .gro file with the .vmd file to make sure it looks as you expect.
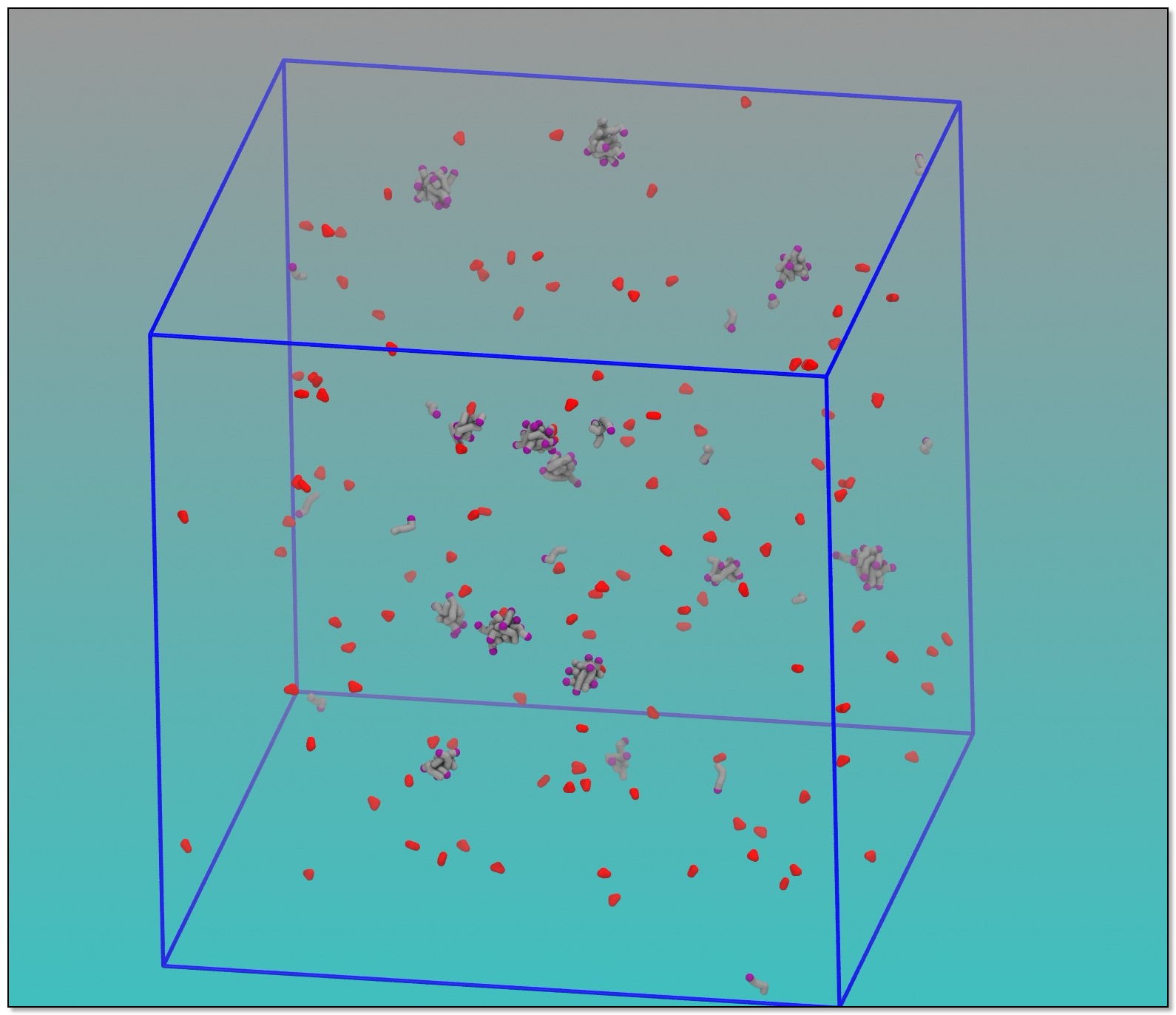
As usual, this process may be automated (for the most part, easily) but putting commands into a file, and running this as a command. Here is an example for the visualization steps: "drymartini.sh".
Suggested workflow for this part — it will help for the remaining bits:
/bin/bash $ cp 1.0mM_System.top 1.0mM_vis.top
(edit 1.0mM_vis.top to take out all but the TTA and BzO/ABZO includes)
/bin/bash $ cp 1.0mM_System.top 1.0mM-run-vis.top
(edit 1.0mM-run-vis.top to remove the last line — the "W" entry)
…proceed with martiniglass process
The TPR file is critical to the analysis, as it contains the bond/connectivity/residue information (analogous to what the simpler PSF file provides in a NAMD run). Unfortunately, VMD is unable to read TPR files created in GROMACS versions beyond 2025.x. Because our main computers are all using GROMACS 2026.x, we have to send some files to a machine with an older version of GROMACS to generate the TPR.
/bin/bash $ rsync -avuz 1.0mM 10.10.160.30:Res
/bin/bash $ ssh 10.10.160.30
/bin/bash (remote) $ cd Res/1.0mM
/bin/bash (remote) $ gmx grompp -f martini_vis_md.mdp -c 10.0mM-run-vis.gro -p 1.0mM-run-vis.top -o 10.0mM-run-vis.tpr
/bin/bash (remote) $ exit
/bin/bash $ scp 10.10.160.30:Res/1.0mM/1.0mM-run-vis.tpr .
Going through these commands line-by-line: we first make sure we are in the (for example) BzO directory, and sync the files from the local directory 1.0mM to the directory Res on the remote computer. Next, we ssh into that computer. After changing to the Res/1.0mM directory, we run gmx grompp to generate our compatible TPR file from our water-less files. Then we log out, and copy that file back to our local computer using scp.
Now we have a TPR file which is compatible with our analysis script. Even though we have run our simulation(s) for 300 ns, it takes a lot of time, at least 100 ns, for the amphiphiles to aggregate. It is good to know what to expect by looking at the energy of the system. Gromacs has a utility for this: gmx energy. This utility will generate data file which may be displayed in a plotting program called xmgrace. (It would be quicker to take a look with GNUplot — see below — but GROMACS makes this more difficult.)
/bin/bash $ gmx energy -f 1.0mM-run.edr -o 1.0mM-run-energy.xmgr
A menu will come up; Enter your energy selections, one per line, and finish with an empty line. I suggest Temperature and Total Energy. This will produce a file with an .xmgr extension. Now open this file in xmgrace:
/bin/bash $ xmgrace 1.0mM-run-energy.xmgr
What are we looking for here? We want to see the total energy, or the temperature, to be stable. That is, we want it to be "flat". xmgrace is a very useful, but complex plotting program, so we will avoid going into too much detail with it here.
We continue to analysis, and we assume that these results files now live in TTA/ABZO/1.0mM, and cmc.tcl lives in TTA (two directory levels above):
/bin/bash $ vmd -dispdev text -e ~/cmc.tcl 1.0mM TTA
The script should run cleanly, with some informational output including the number of frames read, the time in ns, and so on. When it's finished, it will have created two directories and a number of files:
Data-TTA (which contains the aggregate size distributions by mass (Masses.dat) and cluster size (Sizes.dat). These data correspond to the cluster size distributions mentioned above. Until we know which part of our trajectories are useful to look at, we can't really use these data yet.
Cluster-TTA (which holds mostly time-series information)
GNUplot is a program which can allow you to make quick plots of your data from the command line (it can do much more, however!). The plots in the next section were generated in GNUplot. Here is a quick introduction.
Call gnuplot:
/bin/bash $ gnuplot
gnuplot > plot "Cluster-TTA/cluster324.dat" with lines
gnuplot > plot "Cluster-TTA/cluster324.dat" w l
Notice the ability to abbreviate.
gnuplot > plot "Cluster-TTA/cluster324.dat" w lp
"lp" is short for "linespoints" which means lines and points.
gnuplot > plot "Data-TTA/Sizes.dat" using 1:3 with points
"using" (which can be abbreviated as "u") selects columns of data; The first number is the x-, and the second is y-.
Here are some results of a typical simulation. Do you think this system has stabilized? If so, when, and for what time period? These are the questions you want to answer at this stage.
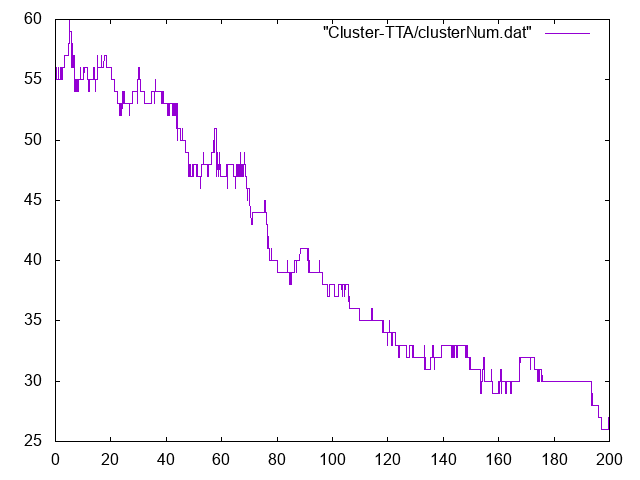
Numbers of clusters vs time
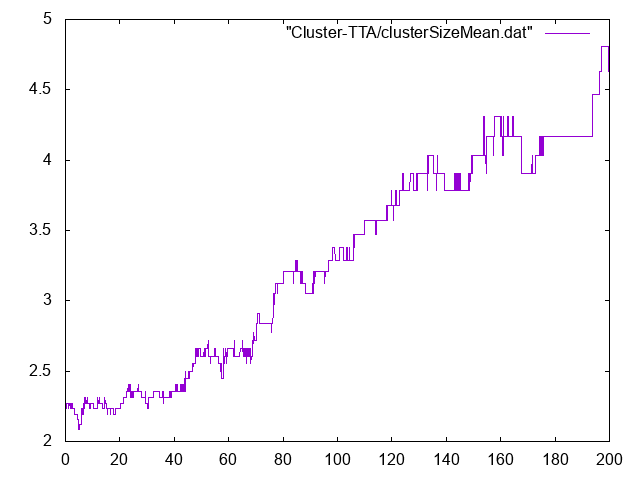
Mean cluster size versus time
Numbers of monomers versus time
Let's assume that your simulation was 300 ns, and after examining the time series data in Cluster-TTA, you decide that the last 100 ns of simulation time are suitable for analysis. You may now use gmx trjconv to create a new trajectory from the original 1.0mM-run-vis.xtc. The -b time switch indicates the time (in ps) to start saving the new trajectory. The number in this example is 100,000 or 100 ns. It is easy to lose track of zeroes!
/bin/bash $ gmx trjconv -f 1.0mM-run-vis.xtc -s 1.0mM-run-vis.tpr -n index.ndx -b 100000 -o 1.0mM-last100-run-vis.xtc
Notice how we have effectively changed the prefix to our data from "1.0mM" to "1.0mM-last100", which will now be our argument to the analysis script. The only problem is that the TPR file does not have the same name. We can fix this by creating a symbolic link to our existing file (instead of simply copying the TPR file to this new name):
/bin/bash $ ln -s 1.0mM-run-vis.tpr 1.0mM-last100-run-vis.tpr
You can make sure it worked the way you expect with an 'ls -ls'. It should look something like this:
/bin/bash $ ls -ls *.tpr
0 lrwxr-xr-x 1 mccallum staff 17 Apr 9 12:39 1.0mM-last100-run-vis.tpr@ -> 1.0mM-run-vis.tpr
72 -rw-r--r-- 1 mccallum staff 36676 Apr 9 12:05 1.0mM-run-vis.tpr
The virtual file is pointing at the real file. Now you can run the analysis script again — this will overwrite all the data in the Cluster-TTA and Data-TTA directories.
/bin/bash $ vmd -dispdev text -e ../../cmc.tcl -args 1.0mM-last100 TTA BzO
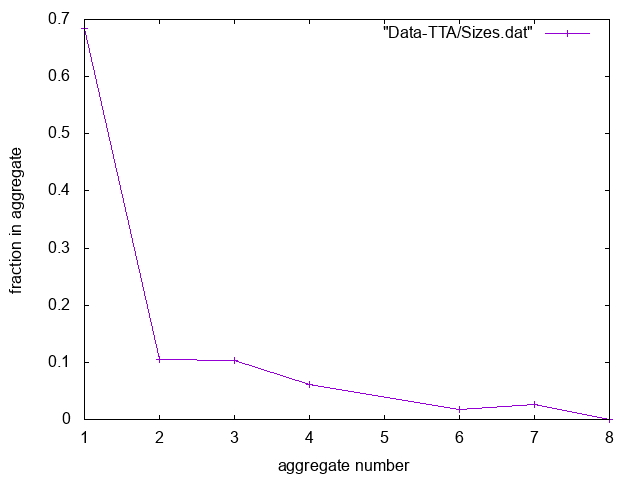
Here is an example of plotting "Sizes.dat". Do you think this is pre- or post-CMC?
Useful commands for trajectories
gmx trjcat -f *.xtc -o concatenated_traj.xtc
PRELUDE: Some background
What are we looking for here? We would like to characterize two conditions: pre-CMC and post-CMC, and hopefully nail down the CMC itself. There are two main measures that a cluster/aggregation analysis can give us (but they are not the only ones!). Essentially, below the CMC, most of the amphiphiles are free monomers. Above the CMC, most of them are in micelles. One way to measure this is by comparing the concentration of free monomers and aggregates versus solution concentration. Free monomer (C1) concentration increases until the CMC, at which point the number remains constant or decreases. The number of amphiphiles in aggregates is small below the CMC, but increases linearly with concentration above the CMC:Monomer and aggregate concentration versus solution concentration
If we can bin the number of amphiphiles in aggregates of size n (Cn) versus n, we should see two behaviors below and above the CMC. Below the CMC, most of the amphiphiles exist as free monomers, so the histogram exponentially (perhaps not literally) decreases from C1 with the fraction close to 1. Above the CMC, there is both a minima in the distribution, and the fraction of monomers is less than 50%:
These things are not directly measurable via experiment, however with simulation we are able to make such an analysis. In the first case, we need to combine measurements from simulations at different concentrations, but with the latter, we can analyze at fixed concentration (per simulation). Both of these analyses will help us fix the CMC according to our model.
PRELUDE: File conventions
It is important to build and follow a consistent file convention. Here is how to do it for this project:
Directory structure: AMPHIPHILE/ANION/CONCENTRATION (that is, each amphiphile will be simulated with a number of anions, and each simulation will be at a different concentration.) For example: TTA/ABZO/1.0mM.
The TTA directory holds all the system martini_v3.0.0 ITP files, along with the TTA ITP and .gro, and water.gro file. The ABZO directory holds the ABZO ITP and .gro files (ABZO_bartender.itp). The simulation directory 1.0mM would have the following files:
- 1.0mM_System.top
- 1.0mM_System.gro
- minimization.mdp
- martini_eq.mdp (equilibrate for 10 ns or 330 000 steps)
- martini_md.mdp (run for 300 ns or 10 000 000 steps)
- run script (optional)
- visualization script (optional)
In addition, you need the analysis script cmc.tcl; This can live anywhere, but it should live in one place, so you don't end up with many (possibly edited) copies, providing inconsistent data between different simulations. I suggest putting it in the TTA directory. Make sure it is executable:
/bin/bash $ chmod 755 cmc.tcl
Step 1: Run your simulation
Step 2: Martiniglass
This is not simply to visualize your system; removing the water from the system reduces the amount of work the analysis script needs to do. You should at least use VMD to look at the .gro file with the .vmd file to make sure it looks as you expect.
As usual, this process may be automated (for the most part, easily) but putting commands into a file, and running this as a command. Here is an example for the visualization steps: "drymartini.sh".
Suggested workflow for this part — it will help for the remaining bits:
/bin/bash $ cp 1.0mM_System.top 1.0mM_vis.top
(edit 1.0mM_vis.top to take out all but the TTA and BzO/ABZO includes)
/bin/bash $ cp 1.0mM_System.top 1.0mM-run-vis.top
(edit 1.0mM-run-vis.top to remove the last line — the "W" entry)
…proceed with martiniglass process
Step 3: Create a compatible TPR and preliminary analysis
The TPR file is critical to the analysis, as it contains the bond/connectivity/residue information (analogous to what the simpler PSF file provides in a NAMD run). Unfortunately, VMD is unable to read TPR files created in GROMACS versions beyond 2025.x. Because our main computers are all using GROMACS 2026.x, we have to send some files to a machine with an older version of GROMACS to generate the TPR.
/bin/bash $ rsync -avuz 1.0mM 10.10.160.30:Res
/bin/bash $ ssh 10.10.160.30
/bin/bash (remote) $ cd Res/1.0mM
/bin/bash (remote) $ gmx grompp -f martini_vis_md.mdp -c 10.0mM-run-vis.gro -p 1.0mM-run-vis.top -o 10.0mM-run-vis.tpr
/bin/bash (remote) $ exit
/bin/bash $ scp 10.10.160.30:Res/1.0mM/1.0mM-run-vis.tpr .
Going through these commands line-by-line: we first make sure we are in the (for example) BzO directory, and sync the files from the local directory 1.0mM to the directory Res on the remote computer. Next, we ssh into that computer. After changing to the Res/1.0mM directory, we run gmx grompp to generate our compatible TPR file from our water-less files. Then we log out, and copy that file back to our local computer using scp.
Now we have a TPR file which is compatible with our analysis script. Even though we have run our simulation(s) for 300 ns, it takes a lot of time, at least 100 ns, for the amphiphiles to aggregate. It is good to know what to expect by looking at the energy of the system. Gromacs has a utility for this: gmx energy. This utility will generate data file which may be displayed in a plotting program called xmgrace. (It would be quicker to take a look with GNUplot — see below — but GROMACS makes this more difficult.)
/bin/bash $ gmx energy -f 1.0mM-run.edr -o 1.0mM-run-energy.xmgr
A menu will come up; Enter your energy selections, one per line, and finish with an empty line. I suggest Temperature and Total Energy. This will produce a file with an .xmgr extension. Now open this file in xmgrace:
/bin/bash $ xmgrace 1.0mM-run-energy.xmgr
What are we looking for here? We want to see the total energy, or the temperature, to be stable. That is, we want it to be "flat". xmgrace is a very useful, but complex plotting program, so we will avoid going into too much detail with it here.
Step 4: The analysis!
We continue to analysis, and we assume that these results files now live in TTA/ABZO/1.0mM, and cmc.tcl lives in TTA (two directory levels above):
- 1.0mM-run-vis.tpr (from our older version of GROMACS)
- 1.0mM-run-vis.xtc
- 1.0mM-run-vis.gro
/bin/bash $ vmd -dispdev text -e ~/cmc.tcl 1.0mM TTA
The script should run cleanly, with some informational output including the number of frames read, the time in ns, and so on. When it's finished, it will have created two directories and a number of files:
Data-TTA (which contains the aggregate size distributions by mass (Masses.dat) and cluster size (Sizes.dat). These data correspond to the cluster size distributions mentioned above. Until we know which part of our trajectories are useful to look at, we can't really use these data yet.
Cluster-TTA (which holds mostly time-series information)
- clusterNum.dat
- clusterSizeMean.dat
- clusterSizeMax.dat
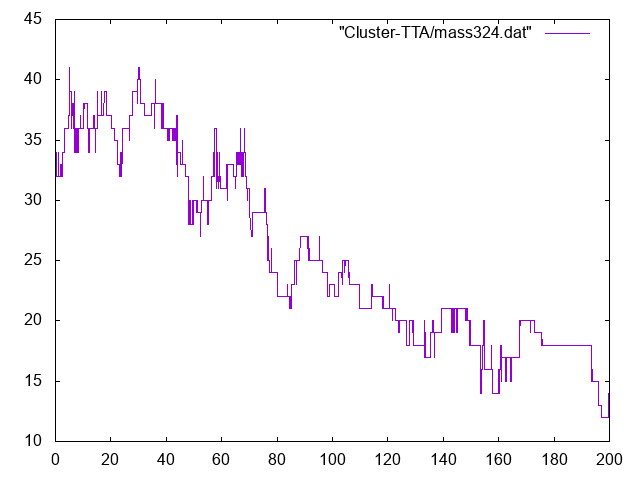
- massXXX.dat
GNUplot
GNUplot is a program which can allow you to make quick plots of your data from the command line (it can do much more, however!). The plots in the next section were generated in GNUplot. Here is a quick introduction.
Call gnuplot:
/bin/bash $ gnuplot
gnuplot > plot "Cluster-TTA/cluster324.dat" with lines
gnuplot > plot "Cluster-TTA/cluster324.dat" w l
Notice the ability to abbreviate.
gnuplot > plot "Cluster-TTA/cluster324.dat" w lp
"lp" is short for "linespoints" which means lines and points.
gnuplot > plot "Data-TTA/Sizes.dat" using 1:3 with points
"using" (which can be abbreviated as "u") selects columns of data; The first number is the x-, and the second is y-.
Time series plots and discussion
Here are some results of a typical simulation. Do you think this system has stabilized? If so, when, and for what time period? These are the questions you want to answer at this stage.
Numbers of clusters vs time
Mean cluster size versus time
Numbers of monomers versus time
Trimming the data and gathering aggregate results
Let's assume that your simulation was 300 ns, and after examining the time series data in Cluster-TTA, you decide that the last 100 ns of simulation time are suitable for analysis. You may now use gmx trjconv to create a new trajectory from the original 1.0mM-run-vis.xtc. The -b time switch indicates the time (in ps) to start saving the new trajectory. The number in this example is 100,000 or 100 ns. It is easy to lose track of zeroes!
/bin/bash $ gmx trjconv -f 1.0mM-run-vis.xtc -s 1.0mM-run-vis.tpr -n index.ndx -b 100000 -o 1.0mM-last100-run-vis.xtc
Notice how we have effectively changed the prefix to our data from "1.0mM" to "1.0mM-last100", which will now be our argument to the analysis script. The only problem is that the TPR file does not have the same name. We can fix this by creating a symbolic link to our existing file (instead of simply copying the TPR file to this new name):
/bin/bash $ ln -s 1.0mM-run-vis.tpr 1.0mM-last100-run-vis.tpr
You can make sure it worked the way you expect with an 'ls -ls'. It should look something like this:
/bin/bash $ ls -ls *.tpr
0 lrwxr-xr-x 1 mccallum staff 17 Apr 9 12:39 1.0mM-last100-run-vis.tpr@ -> 1.0mM-run-vis.tpr
72 -rw-r--r-- 1 mccallum staff 36676 Apr 9 12:05 1.0mM-run-vis.tpr
The virtual file is pointing at the real file. Now you can run the analysis script again — this will overwrite all the data in the Cluster-TTA and Data-TTA directories.
/bin/bash $ vmd -dispdev text -e ../../cmc.tcl -args 1.0mM-last100 TTA BzO
Results of the analysis
Here is an example of plotting "Sizes.dat". Do you think this is pre- or post-CMC?
Useful commands for trajectories
gmx trjcat -f *.xtc -o concatenated_traj.xtc